 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Many-Body Expansion Combined with Neural Networks
Paper and Code
Sep 22, 2016
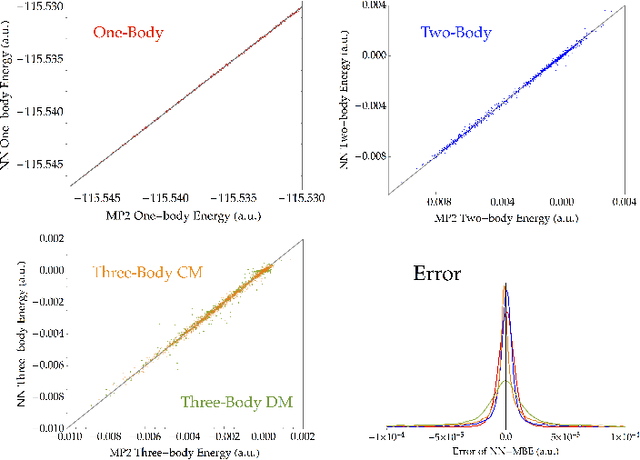
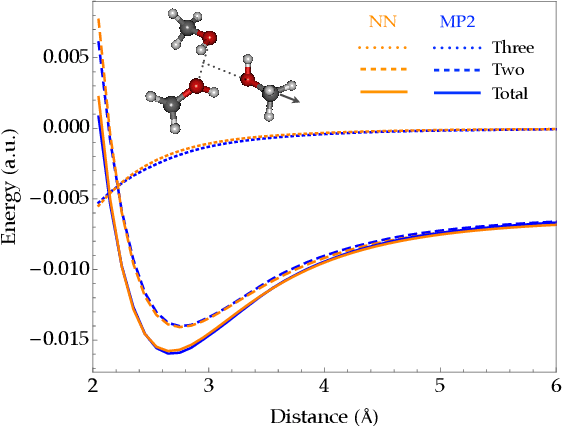
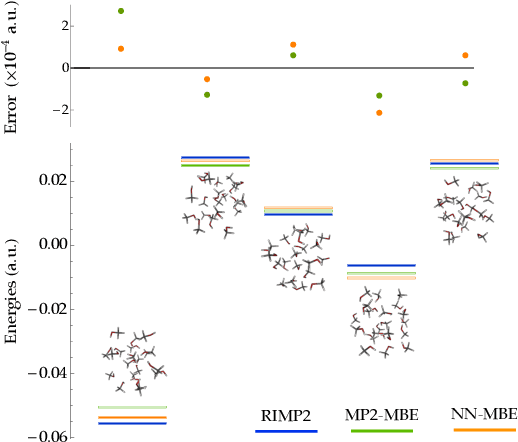
Fragmentation methods such as the many-body expansion (MBE) are a common strategy to model large systems by partitioning energies into a hierarchy of decreasingly significant contributions. The number of fragments required for chemical accuracy is still prohibitively expensive for ab-initio MBE to compete with force field approximations for applications beyond single-point energies. Alongside the MBE, empirical models of ab-initio potential energy surfaces have improved, especially non-linear models based on neural networks (NN) which can reproduce ab-initio potential energy surfaces rapidly and accurately. Although they are fast, NNs suffer from their own curse of dimensionality; they must be trained on a representative sample of chemical space. In this paper we examine the synergy of the MBE and NN's, and explore their complementarity. The MBE offers a systematic way to treat systems of arbitrary size and intelligently sample chemical space. NN's reduce, by a factor in excess of $10^6$ the computational overhead of the MBE and reproduce the accuracy of ab-initio calculations without specialized force fields. We show they are remarkably general, providing comparable accuracy with drastically different chemical embeddings. To assess this we test a new chemical embedding which can be inverted to predict molecules with desired properties.