 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBayesian Metabolic Flux Analysis reveals intracellular flux couplings
Apr 18, 2018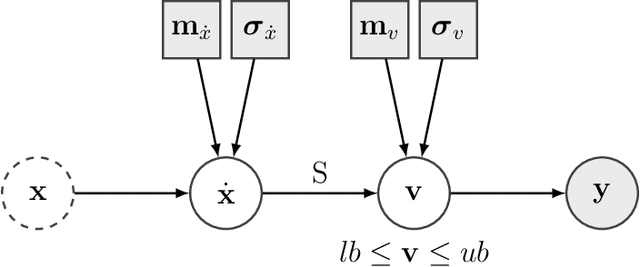
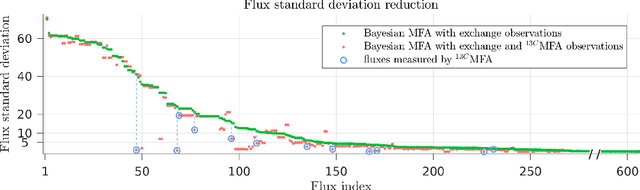
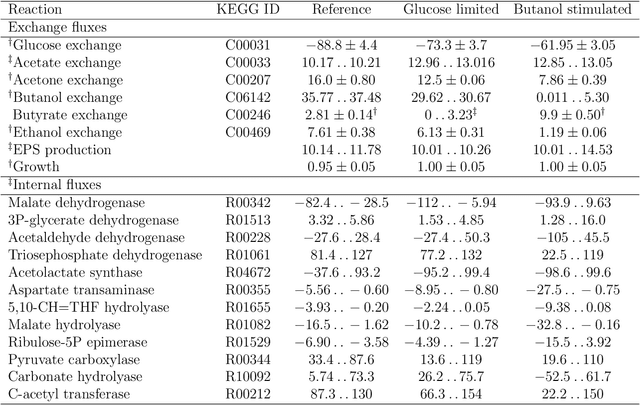
Metabolic flux balance analyses are a standard tool in analysing metabolic reaction rates compatible with measurements, steady-state and the metabolic reaction network stoichiometry. Flux analysis methods commonly place unrealistic assumptions on fluxes due to the convenience of formulating the problem as a linear programming model, and most methods ignore the notable uncertainty in flux estimates. We introduce a novel paradigm of Bayesian metabolic flux analysis that models the reactions of the whole genome-scale cellular system in probabilistic terms, and can infer the full flux vector distribution of genome-scale metabolic systems based on exchange and intracellular (e.g. 13C) flux measurements, steady-state assumptions, and target function assumptions. The Bayesian model couples all fluxes jointly together in a simple truncated multivariate posterior distribution, which reveals informative flux couplings. Our model is a plug-in replacement to conventional metabolic balance methods, such as flux balance analysis (FBA). Our experiments indicate that we can characterise the genome-scale flux covariances, reveal flux couplings, and determine more intracellular unobserved fluxes in C. acetobutylicum from 13C data than flux variability analysis. The COBRA compatible software is available at github.com/markusheinonen/bamfa