 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEmerging Microelectronic Materials by Design: Navigating Combinatorial Design Space with Scarce and Dispersed Data
Dec 23, 2024



The increasing demands of sustainable energy, electronics, and biomedical applications call for next-generation functional materials with unprecedented properties. Of particular interest are emerging materials that display exceptional physical properties, making them promising candidates in energy-efficient microelectronic devices. As the conventional Edisonian approach becomes significantly outpaced by growing societal needs, emerging computational modeling and machine learning (ML) methods are employed for the rational design of materials. However, the complex physical mechanisms, cost of first-principles calculations, and the dispersity and scarcity of data pose challenges to both physics-based and data-driven materials modeling. Moreover, the combinatorial composition-structure design space is high-dimensional and often disjoint, making design optimization nontrivial. In this Account, we review a team effort toward establishing a framework that integrates data-driven and physics-based methods to address these challenges and accelerate materials design. We begin by presenting our integrated materials design framework and its three components in a general context. We then provide an example of applying this materials design framework to metal-insulator transition (MIT) materials, a specific type of emerging materials with practical importance in next-generation memory technologies. We identify multiple new materials which may display this property and propose pathways for their synthesis. Finally, we identify some outstanding challenges in data-driven materials design, such as materials data quality issues and property-performance mismatch. We seek to raise awareness of these overlooked issues hindering materials design, thus stimulating efforts toward developing methods to mitigate the gaps.
Do Graph Neural Networks Work for High Entropy Alloys?
Aug 29, 2024

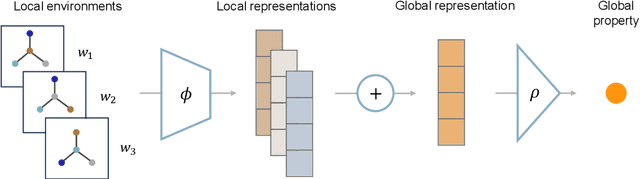
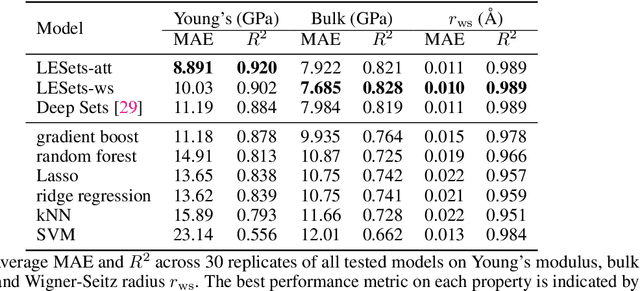
Graph neural networks (GNNs) have excelled in predictive modeling for both crystals and molecules, owing to the expressiveness of graph representations. High-entropy alloys (HEAs), however, lack chemical long-range order, limiting the applicability of current graph representations. To overcome this challenge, we propose a representation of HEAs as a collection of local environment (LE) graphs. Based on this representation, we introduce the LESets machine learning model, an accurate, interpretable GNN for HEA property prediction. We demonstrate the accuracy of LESets in modeling the mechanical properties of quaternary HEAs. Through analyses and interpretation, we further extract insights into the modeling and design of HEAs. In a broader sense, LESets extends the potential applicability of GNNs to disordered materials with combinatorial complexity formed by diverse constituents and their flexible configurations.
MolSets: Molecular Graph Deep Sets Learning for Mixture Property Modeling
Dec 27, 2023Recent advances in machine learning (ML) have expedited materials discovery and design. One significant challenge faced in ML for materials is the expansive combinatorial space of potential materials formed by diverse constituents and their flexible configurations. This complexity is particularly evident in molecular mixtures, a frequently explored space for materials such as battery electrolytes. Owing to the complex structures of molecules and the sequence-independent nature of mixtures, conventional ML methods have difficulties in modeling such systems. Here we present MolSets, a specialized ML model for molecular mixtures. Representing individual molecules as graphs and their mixture as a set, MolSets leverages a graph neural network and the deep sets architecture to extract information at the molecule level and aggregate it at the mixture level, thus addressing local complexity while retaining global flexibility. We demonstrate the efficacy of MolSets in predicting the conductivity of lithium battery electrolytes and highlight its benefits in virtual screening of the combinatorial chemical space.
ET-AL: Entropy-Targeted Active Learning for Bias Mitigation in Materials Data
Dec 07, 2022



Growing materials data and data-driven informatics drastically promote the discovery and design of materials. While there are significant advancements in data-driven models, the quality of data resources is less studied despite its huge impact on model performance. In this work, we focus on data bias arising from uneven coverage of materials families in existing knowledge. Observing different diversities among crystal systems in common materials databases, we propose an information entropy-based metric for measuring this bias. To mitigate the bias, we develop an entropy-targeted active learning (ET-AL) framework, which guides the acquisition of new data to improve the diversity of underrepresented crystal systems. We demonstrate the capability of ET-AL for bias mitigation and the resulting improvement in downstream machine learning models. This approach is broadly applicable to data-driven materials discovery, including autonomous data acquisition and dataset trimming to reduce bias, as well as data-driven informatics in other scientific domains.