 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtSCAPE: Mapping the landscape of protein conformations in molecular dynamics
Oct 27, 2024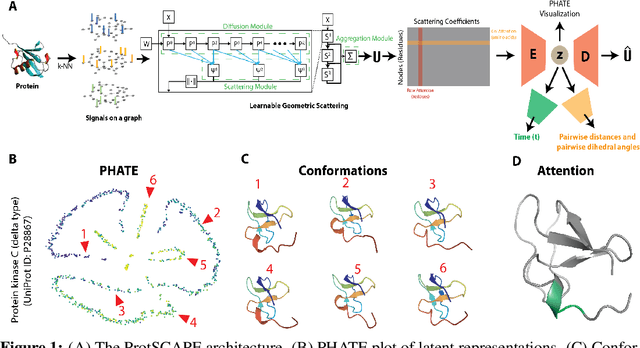
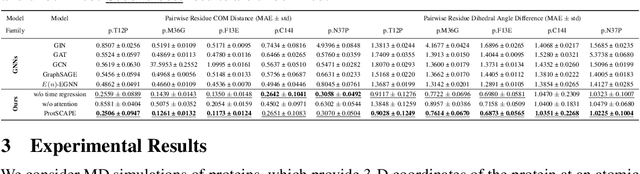
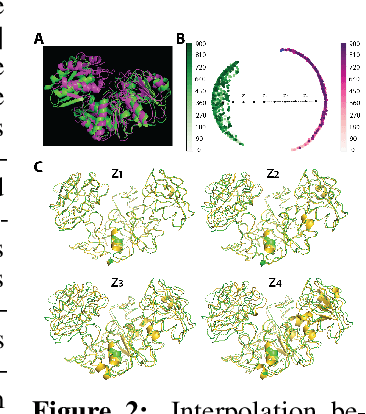
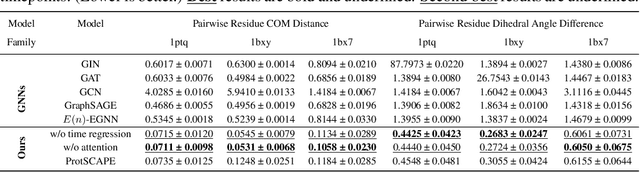
Understanding the dynamic nature of protein structures is essential for comprehending their biological functions. While significant progress has been made in predicting static folded structures, modeling protein motions on microsecond to millisecond scales remains challenging. To address these challenges, we introduce a novel deep learning architecture, Protein Transformer with Scattering, Attention, and Positional Embedding (ProtSCAPE), which leverages the geometric scattering transform alongside transformer-based attention mechanisms to capture protein dynamics from molecular dynamics (MD) simulations. ProtSCAPE utilizes the multi-scale nature of the geometric scattering transform to extract features from protein structures conceptualized as graphs and integrates these features with dual attention structures that focus on residues and amino acid signals, generating latent representations of protein trajectories. Furthermore, ProtSCAPE incorporates a regression head to enforce temporally coherent latent representations.
Molecular Graph Generation via Geometric Scattering
Oct 12, 2021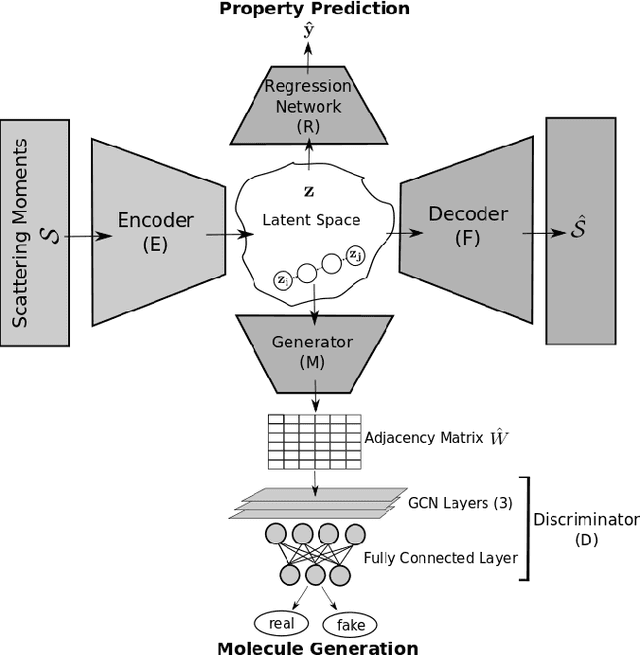
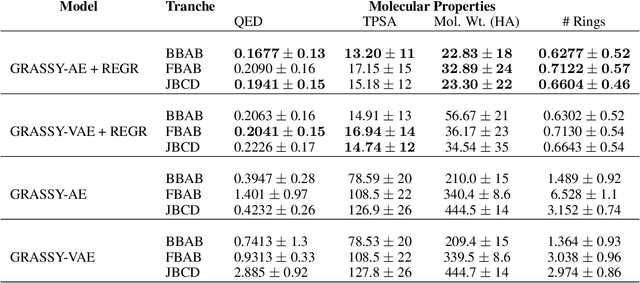

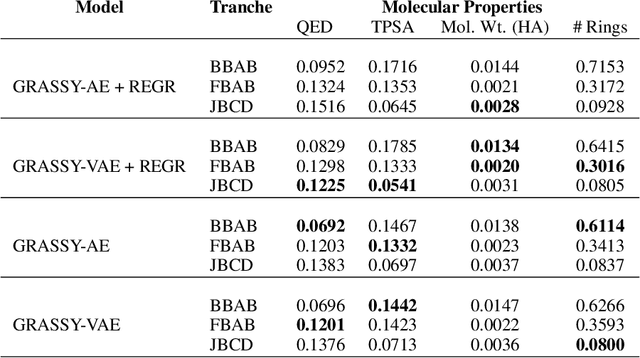
Graph neural networks (GNNs) have been used extensively for addressing problems in drug design and discovery. Both ligand and target molecules are represented as graphs with node and edge features encoding information about atomic elements and bonds respectively. Although existing deep learning models perform remarkably well at predicting physicochemical properties and binding affinities, the generation of new molecules with optimized properties remains challenging. Inherently, most GNNs perform poorly in whole-graph representation due to the limitations of the message-passing paradigm. Furthermore, step-by-step graph generation frameworks that use reinforcement learning or other sequential processing can be slow and result in a high proportion of invalid molecules with substantial post-processing needed in order to satisfy the principles of stoichiometry. To address these issues, we propose a representation-first approach to molecular graph generation. We guide the latent representation of an autoencoder by capturing graph structure information with the geometric scattering transform and apply penalties that structure the representation also by molecular properties. We show that this highly structured latent space can be directly used for molecular graph generation by the use of a GAN. We demonstrate that our architecture learns meaningful representations of drug datasets and provides a platform for goal-directed drug synthesis.