 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSampling 3D Molecular Conformers with Diffusion Transformers
Jun 18, 2025



Diffusion Transformers (DiTs) have demonstrated strong performance in generative modeling, particularly in image synthesis, making them a compelling choice for molecular conformer generation. However, applying DiTs to molecules introduces novel challenges, such as integrating discrete molecular graph information with continuous 3D geometry, handling Euclidean symmetries, and designing conditioning mechanisms that generalize across molecules of varying sizes and structures. We propose DiTMC, a framework that adapts DiTs to address these challenges through a modular architecture that separates the processing of 3D coordinates from conditioning on atomic connectivity. To this end, we introduce two complementary graph-based conditioning strategies that integrate seamlessly with the DiT architecture. These are combined with different attention mechanisms, including both standard non-equivariant and SO(3)-equivariant formulations, enabling flexible control over the trade-off between between accuracy and computational efficiency. Experiments on standard conformer generation benchmarks (GEOM-QM9, -DRUGS, -XL) demonstrate that DiTMC achieves state-of-the-art precision and physical validity. Our results highlight how architectural choices and symmetry priors affect sample quality and efficiency, suggesting promising directions for large-scale generative modeling of molecular structures. Code available at https://github.com/ML4MolSim/dit_mc.
Euclidean Fast Attention: Machine Learning Global Atomic Representations at Linear Cost
Dec 11, 2024Long-range correlations are essential across numerous machine learning tasks, especially for data embedded in Euclidean space, where the relative positions and orientations of distant components are often critical for accurate predictions. Self-attention offers a compelling mechanism for capturing these global effects, but its quadratic complexity presents a significant practical limitation. This problem is particularly pronounced in computational chemistry, where the stringent efficiency requirements of machine learning force fields (MLFFs) often preclude accurately modeling long-range interactions. To address this, we introduce Euclidean fast attention (EFA), a linear-scaling attention-like mechanism designed for Euclidean data, which can be easily incorporated into existing model architectures. A core component of EFA are novel Euclidean rotary positional encodings (ERoPE), which enable efficient encoding of spatial information while respecting essential physical symmetries. We empirically demonstrate that EFA effectively captures diverse long-range effects, enabling EFA-equipped MLFFs to describe challenging chemical interactions for which conventional MLFFs yield incorrect results.
From Peptides to Nanostructures: A Euclidean Transformer for Fast and Stable Machine Learned Force Fields
Sep 21, 2023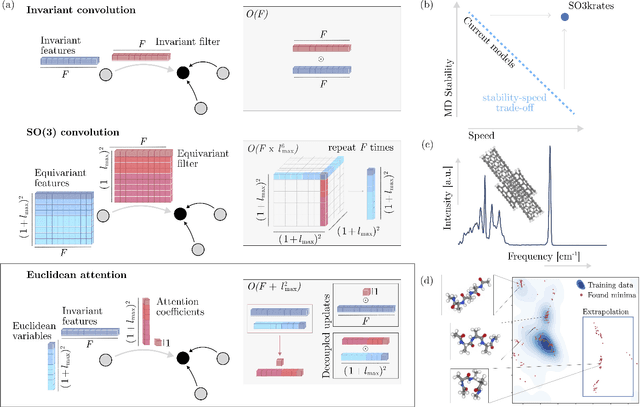
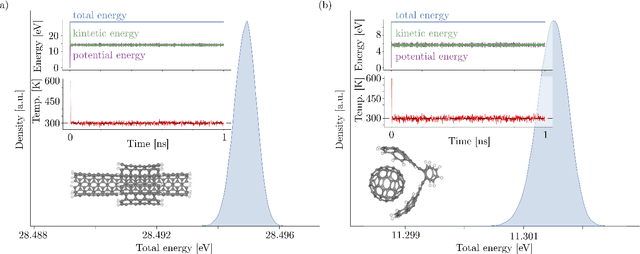
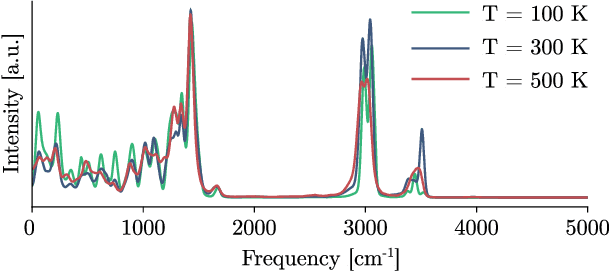
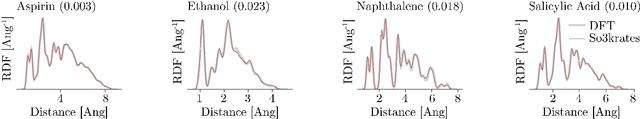
Recent years have seen vast progress in the development of machine learned force fields (MLFFs) based on ab-initio reference calculations. Despite achieving low test errors, the suitability of MLFFs in molecular dynamics (MD) simulations is being increasingly scrutinized due to concerns about instability. Our findings suggest a potential connection between MD simulation stability and the presence of equivariant representations in MLFFs, but their computational cost can limit practical advantages they would otherwise bring. To address this, we propose a transformer architecture called SO3krates that combines sparse equivariant representations (Euclidean variables) with a self-attention mechanism that can separate invariant and equivariant information, eliminating the need for expensive tensor products. SO3krates achieves a unique combination of accuracy, stability, and speed that enables insightful analysis of quantum properties of matter on unprecedented time and system size scales. To showcase this capability, we generate stable MD trajectories for flexible peptides and supra-molecular structures with hundreds of atoms. Furthermore, we investigate the PES topology for medium-sized chainlike molecules (e.g., small peptides) by exploring thousands of minima. Remarkably, SO3krates demonstrates the ability to strike a balance between the conflicting demands of stability and the emergence of new minimum-energy conformations beyond the training data, which is crucial for realistic exploration tasks in the field of biochemistry.
Stress and heat flux via automatic differentiation
May 02, 2023Machine-learning potentials provide computationally efficient and accurate approximations of the Born-Oppenheimer potential energy surface. This potential determines many materials properties and simulation techniques usually require its gradients, in particular forces and stress for molecular dynamics, and heat flux for thermal transport properties. Recently developed potentials feature high body order and can include equivariant semi-local interactions through message-passing mechanisms. Due to their complex functional forms, they rely on automatic differentiation (AD), overcoming the need for manual implementations or finite-difference schemes to evaluate gradients. This study demonstrates a unified AD approach to obtain forces, stress, and heat flux for such potentials, and provides a model-independent implementation. The method is tested on the Lennard-Jones potential, and then applied to predict cohesive properties and thermal conductivity of tin selenide using an equivariant message-passing neural network potential.
So3krates -- Self-attention for higher-order geometric interactions on arbitrary length-scales
May 28, 2022

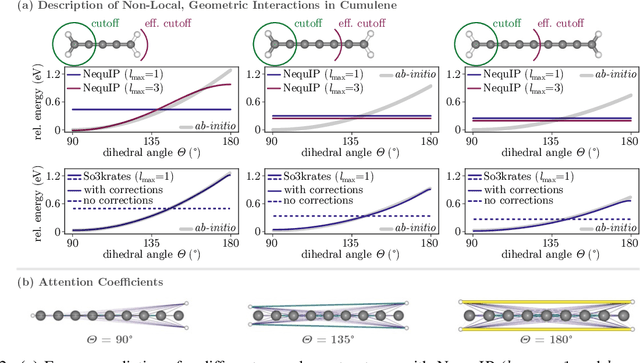

The application of machine learning methods in quantum chemistry has enabled the study of numerous chemical phenomena, which are computationally intractable with traditional ab-initio methods. However, some quantum mechanical properties of molecules and materials depend on non-local electronic effects, which are often neglected due to the difficulty of modeling them efficiently. This work proposes a modified attention mechanism adapted to the underlying physics, which allows to recover the relevant non-local effects. Namely, we introduce spherical harmonic coordinates (SPHCs) to reflect higher-order geometric information for each atom in a molecule, enabling a non-local formulation of attention in the SPHC space. Our proposed model So3krates -- a self-attention based message passing neural network -- uncouples geometric information from atomic features, making them independently amenable to attention mechanisms. We show that in contrast to other published methods, So3krates is able to describe non-local quantum mechanical effects over arbitrary length scales. Further, we find evidence that the inclusion of higher-order geometric correlations increases data efficiency and improves generalization. So3krates matches or exceeds state-of-the-art performance on popular benchmarks, notably, requiring a significantly lower number of parameters (0.25--0.4x) while at the same time giving a substantial speedup (6--14x for training and 2--11x for inference) compared to other models.