 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDiagnosing Heteroskedasticity and Resolving Multicollinearity Paradoxes in Physicochemical Property Prediction
Jan 01, 2026Lipophilicity (logP) prediction remains central to drug discovery, yet linear regression models for this task frequently violate statistical assumptions in ways that invalidate their reported performance metrics. We analyzed 426,850 bioactive molecules from a rigorously curated intersection of PubChem, ChEMBL, and eMolecules databases, revealing severe heteroskedasticity in linear models predicting computed logP values (XLOGP3): residual variance increases 4.2-fold in lipophilic regions (logP greater than 5) compared to balanced regions (logP 2 to 4). Classical remediation strategies (Weighted Least Squares and Box-Cox transformation) failed to resolve this violation (Breusch-Pagan p-value less than 0.0001 for all variants). Tree-based ensemble methods (Random Forest R-squared of 0.764, XGBoost R-squared of 0.765) proved inherently robust to heteroskedasticity while delivering superior predictive performance. SHAP analysis resolved a critical multicollinearity paradox: despite a weak bivariate correlation of 0.146, molecular weight emerged as the single most important predictor (mean absolute SHAP value of 0.573), with its effect suppressed in simple correlations by confounding with topological polar surface area (TPSA). These findings demonstrate that standard linear models face fundamental challenges for computed lipophilicity prediction and provide a principled framework for interpreting ensemble models in QSAR applications.
A Scalable Framework for logP Prediction: From Terabyte-Scale Data Integration to Interpretable Ensemble Modeling
Dec 31, 2025This study presents a large-scale predictive modeling framework for logP prediction using 426850 bioactive compounds rigorously curated from the intersection of three authoritative chemical databases: PubChem, ChEMBL, and eMolecules. We developed a novel computational infrastructure to address the data integration challenge, reducing processing time from a projected over 100 days to 3.2 hours through byte-offset indexing architecture, a 740-fold improvement. Our comprehensive analysis revealed critical insights into the multivariate nature of lipophilicity: while molecular weight exhibited weak bivariate correlation with logP, SHAP analysis on ensemble models identified it as the single most important predictor globally. We systematically evaluated multiple modeling approaches, discovering that linear models suffered from inherent heteroskedasticity that classical remediation strategies, including weighted least squares and Box-Cox transformation, failed to address. Tree-based ensemble methods, including Random Forest and XGBoost, proved inherently robust to this violation, achieving an R-squared of 0.765 and RMSE of 0.731 logP units on the test set. Furthermore, a stratified modeling strategy, employing specialized models for drug-like molecules (91 percent of dataset) and extreme cases (nine percent), achieved optimal performance: an RMSE of 0.838 for the drug-like subset and an R-squared of 0.767 for extreme molecules, the highest of all evaluated approaches. These findings provide actionable guidance for molecular design, establish robust baselines for lipophilicity prediction using only 2D descriptors, and demonstrate that well-curated, descriptor-based ensemble models remain competitive with state-of-the-art graph neural network architectures.
Bridging the Plausibility-Validity Gap by Fine-Tuning a Reasoning-Enhanced LLM for Chemical Synthesis and Discovery
Jul 09, 2025Large Language Models (LLMs) often generate scientifically plausible but factually invalid information, a challenge we term the "plausibility-validity gap," particularly in specialized domains like chemistry. This paper presents a systematic methodology to bridge this gap by developing a specialized scientific assistant. We utilized the Magistral Small model, noted for its integrated reasoning capabilities, and fine-tuned it using Low-Rank Adaptation (LoRA). A key component of our approach was the creation of a "dual-domain dataset," a comprehensive corpus curated from various sources encompassing both molecular properties and chemical reactions, which was standardized to ensure quality. Our evaluation demonstrates that the fine-tuned model achieves significant improvements over the baseline model in format adherence, chemical validity of generated molecules, and the feasibility of proposed synthesis routes. The results indicate a hierarchical learning pattern, where syntactic correctness is learned more readily than chemical possibility and synthesis feasibility. While a comparative analysis with human experts revealed competitive performance in areas like chemical creativity and reasoning, it also highlighted key limitations, including persistent errors in stereochemistry, a static knowledge cutoff, and occasional reference hallucination. This work establishes a viable framework for adapting generalist LLMs into reliable, specialized tools for chemical research, while also delineating critical areas for future improvement.
NAPER: Fault Protection for Real-Time Resource-Constrained Deep Neural Networks
Apr 09, 2025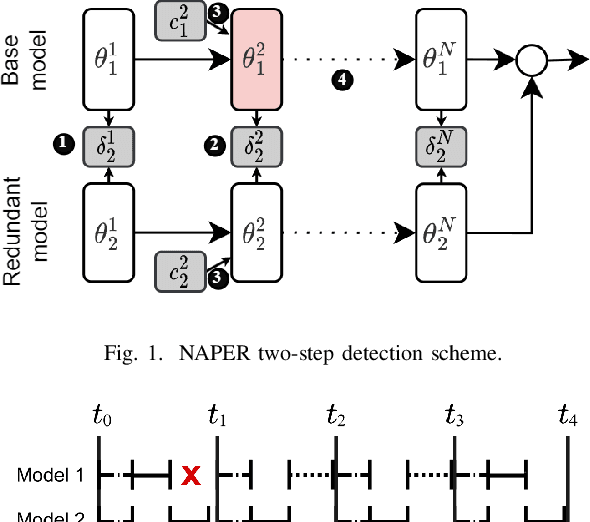
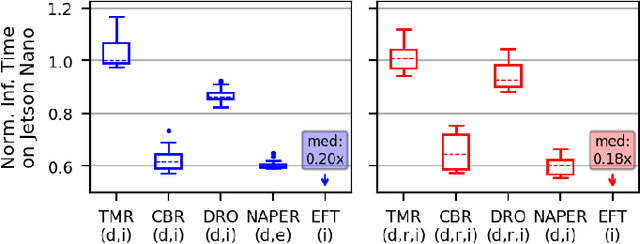
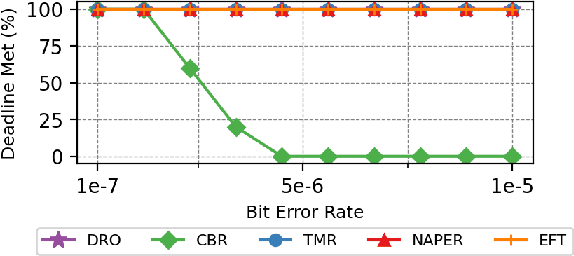
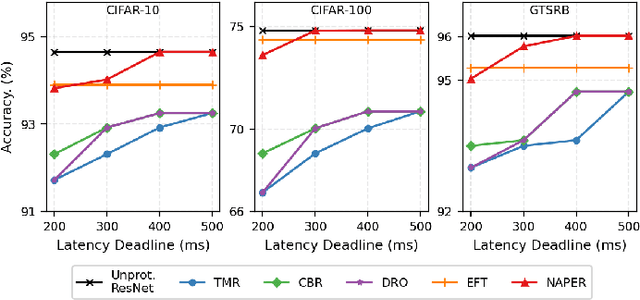
Fault tolerance in Deep Neural Networks (DNNs) deployed on resource-constrained systems presents unique challenges for high-accuracy applications with strict timing requirements. Memory bit-flips can severely degrade DNN accuracy, while traditional protection approaches like Triple Modular Redundancy (TMR) often sacrifice accuracy to maintain reliability, creating a three-way dilemma between reliability, accuracy, and timeliness. We introduce NAPER, a novel protection approach that addresses this challenge through ensemble learning. Unlike conventional redundancy methods, NAPER employs heterogeneous model redundancy, where diverse models collectively achieve higher accuracy than any individual model. This is complemented by an efficient fault detection mechanism and a real-time scheduler that prioritizes meeting deadlines by intelligently scheduling recovery operations without interrupting inference. Our evaluations demonstrate NAPER's superiority: 40% faster inference in both normal and fault conditions, maintained accuracy 4.2% higher than TMR-based strategies, and guaranteed uninterrupted operation even during fault recovery. NAPER effectively balances the competing demands of accuracy, reliability, and timeliness in real-time DNN applications