 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNACNet: A Histology Context-aware Transformer Graph Convolution Network for Predicting Treatment Response to Neoadjuvant Chemotherapy in Triple Negative Breast Cancer
Nov 14, 2024



Neoadjuvant chemotherapy (NAC) response prediction for triple negative breast cancer (TNBC) patients is a challenging task clinically as it requires understanding complex histology interactions within the tumor microenvironment (TME). Digital whole slide images (WSIs) capture detailed tissue information, but their giga-pixel size necessitates computational methods based on multiple instance learning, which typically analyze small, isolated image tiles without the spatial context of the TME. To address this limitation and incorporate TME spatial histology interactions in predicting NAC response for TNBC patients, we developed a histology context-aware transformer graph convolution network (NACNet). Our deep learning method identifies the histopathological labels on individual image tiles from WSIs, constructs a spatial TME graph, and represents each node with features derived from tissue texture and social network analysis. It predicts NAC response using a transformer graph convolution network model enhanced with graph isomorphism network layers. We evaluate our method with WSIs of a cohort of TNBC patient (N=105) and compared its performance with multiple state-of-the-art machine learning and deep learning models, including both graph and non-graph approaches. Our NACNet achieves 90.0% accuracy, 96.0% sensitivity, 88.0% specificity, and an AUC of 0.82, through eight-fold cross-validation, outperforming baseline models. These comprehensive experimental results suggest that NACNet holds strong potential for stratifying TNBC patients by NAC response, thereby helping to prevent overtreatment, improve patient quality of life, reduce treatment cost, and enhance clinical outcomes, marking an important advancement toward personalized breast cancer treatment.
Clumped Nuclei Segmentation with Adjacent Point Match and Local Shape based Intensity Analysis for Overlapped Nuclei in Fluorescence In-Situ Hybridization Images
Aug 14, 2018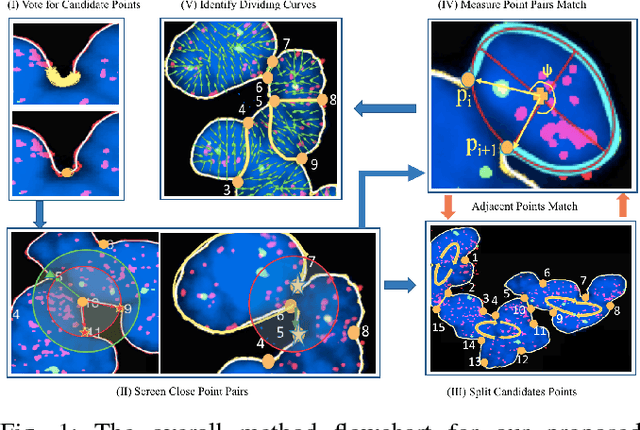
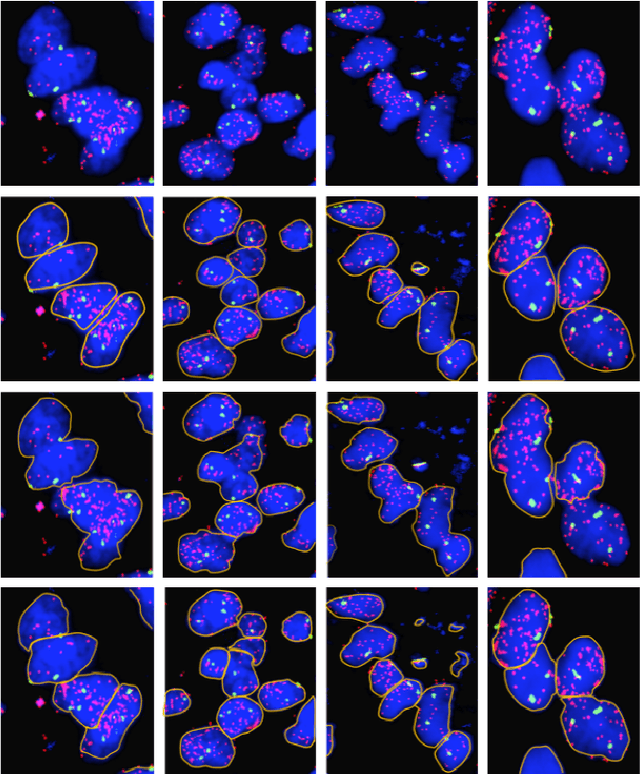
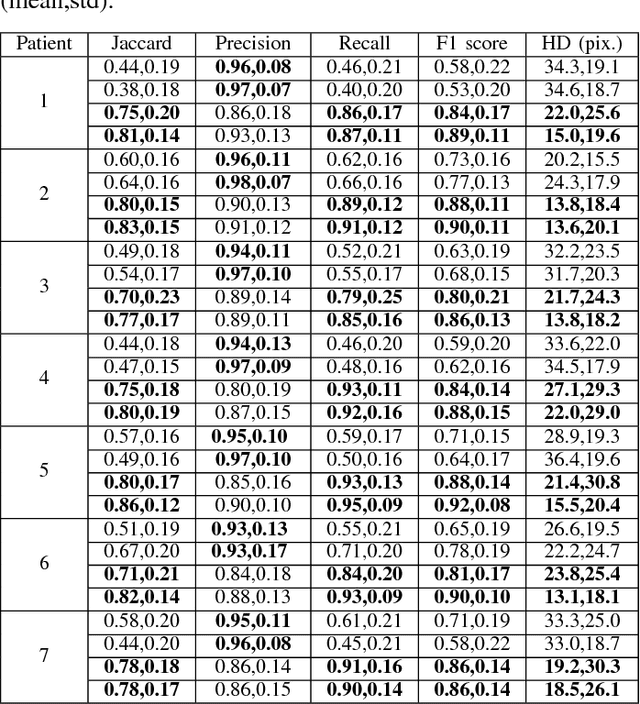
Highly clumped nuclei clusters captured in fluorescence in situ hybridization microscopy images are common histology entities under investigations in a wide spectrum of tissue-related biomedical investigations. Due to their large scale in presence, computer based image analysis is used to facilitate such analysis with improved analysis efficiency and reproducibility. To ensure the quality of downstream biomedical analyses, it is essential to segment clustered nuclei with high quality. However, this presents a technical challenge commonly encountered in a large number of biomedical research, as nuclei are often overlapped due to a high cell density. In this paper, we propose an segmentation algorithm that identifies point pair connection candidates and evaluates adjacent point connections with a formulated ellipse fitting quality indicator. After connection relationships are determined, we recover the resulting dividing paths by following points with specific eigenvalues from Hessian in a constrained searching space. We validate our algorithm with 560 image patches from two classes of tumor regions of seven brain tumor patients. Both qualitative and quantitative experimental results suggest that our algorithm is promising for dividing overlapped nuclei in fluorescence in situ hybridization microscopy images widely used in various biomedical research.
Analysis of Cellular Feature Differences of Astrocytomas with Distinct Mutational Profiles Using Digitized Histopathology Images
Jun 24, 2018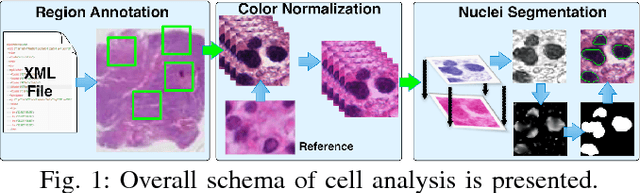
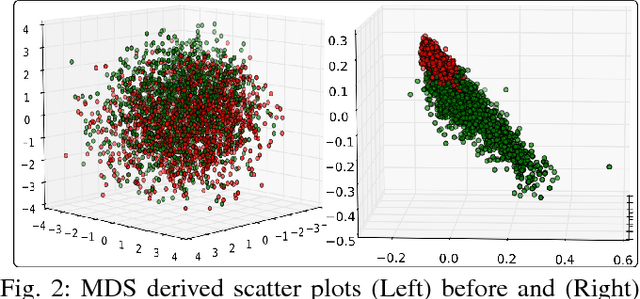
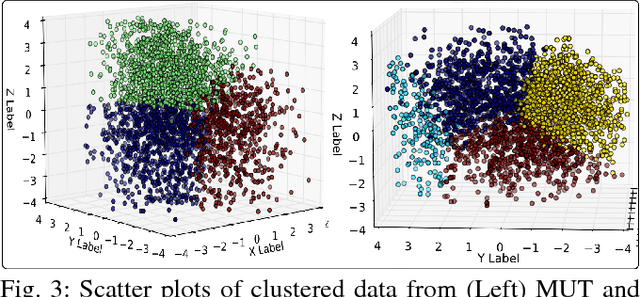
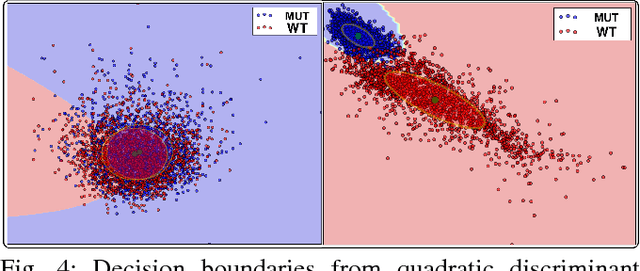
Cellular phenotypic features derived from histopathology images are the basis of pathologic diagnosis and are thought to be related to underlying molecular profiles. Due to overwhelming cell numbers and population heterogeneity, it remains challenging to quantitatively compute and compare features of cells with distinct molecular signatures. In this study, we propose a self-reliant and efficient analysis framework that supports quantitative analysis of cellular phenotypic difference across distinct molecular groups. To demonstrate efficacy, we quantitatively analyze astrocytomas that are molecularly characterized as either Isocitrate Dehydrogenase (IDH) mutant (MUT) or wildtype (WT) using imaging data from The Cancer Genome Atlas database. Representative cell instances that are phenotypically different between these two groups are retrieved after segmentation, feature computation, data pruning, dimensionality reduction, and unsupervised clustering. Our analysis is generic and can be applied to a wide set of cell-based biomedical research.
Segmentation of Overlapped Steatosis in Whole-Slide Liver Histopathology Microscopy Images
Jun 24, 2018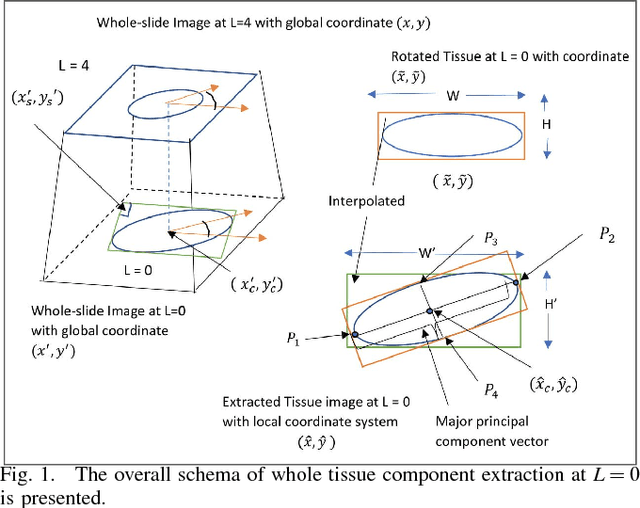
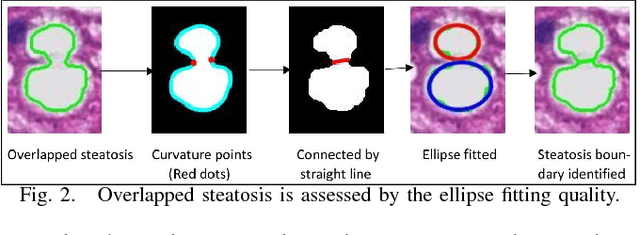
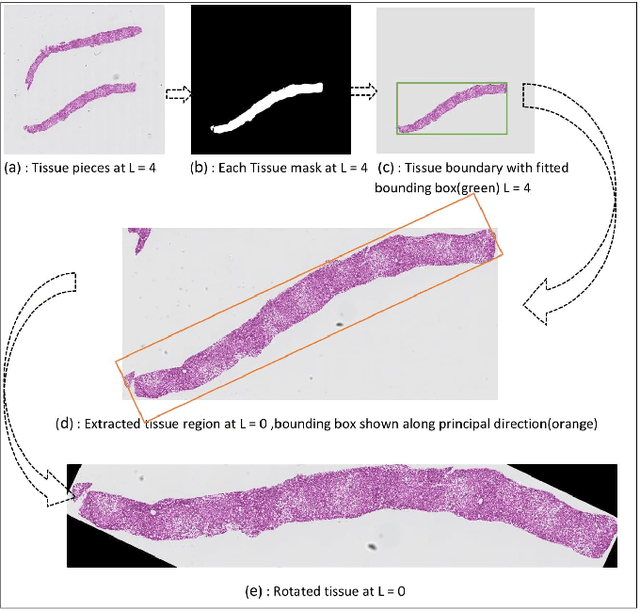
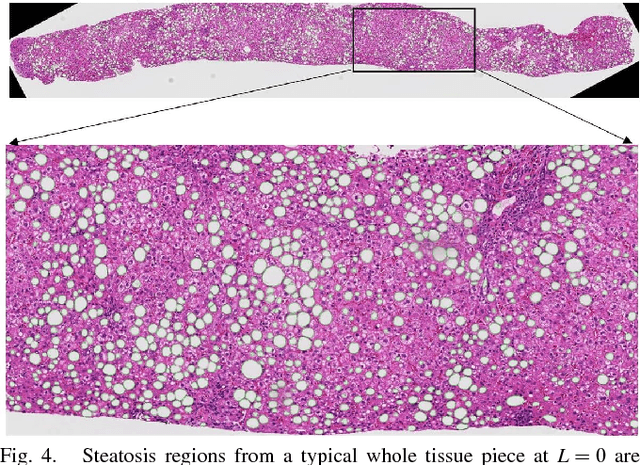
An accurate steatosis quantification with pathology tissue samples is of high clinical importance. However, such pathology measurement is manually made in most clinical practices, subject to severe reader variability due to large sampling bias and poor reproducibility. Although some computerized automated methods are developed to quantify the steatosis regions, they present limited analysis capacity for high resolution whole-slide microscopy images and accurate overlapped steatosis division. In this paper, we propose a method that extracts an individual whole tissue piece at high resolution with minimum background area by estimating tissue bounding box and rotation angle. This is followed by the segmentation and segregation of steatosis regions with high curvature point detection and an ellipse fitting quality assessment method. We validate our method with isolated and overlapped steatosis regions in liver tissue images of 11 patients. The experimental results suggest that our method is promising for enhanced support of steatosis quantization during the pathology review for liver disease treatment.