 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Potential Energy Surfaces of Hydrogen Atom Transfer Reactions in Peptides
Aug 01, 2025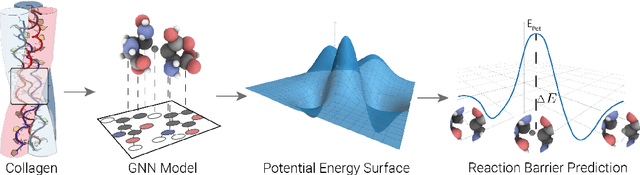
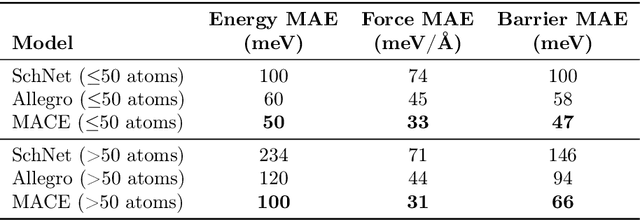
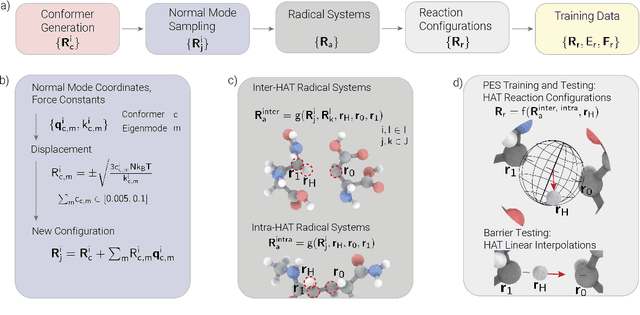

Hydrogen atom transfer (HAT) reactions are essential in many biological processes, such as radical migration in damaged proteins, but their mechanistic pathways remain incompletely understood. Simulating HAT is challenging due to the need for quantum chemical accuracy at biologically relevant scales; thus, neither classical force fields nor DFT-based molecular dynamics are applicable. Machine-learned potentials offer an alternative, able to learn potential energy surfaces (PESs) with near-quantum accuracy. However, training these models to generalize across diverse HAT configurations, especially at radical positions in proteins, requires tailored data generation and careful model selection. Here, we systematically generate HAT configurations in peptides to build large datasets using semiempirical methods and DFT. We benchmark three graph neural network architectures (SchNet, Allegro, and MACE) on their ability to learn HAT PESs and indirectly predict reaction barriers from energy predictions. MACE consistently outperforms the others in energy, force, and barrier prediction, achieving a mean absolute error of 1.13 kcal/mol on out-of-distribution DFT barrier predictions. This accuracy enables integration of ML potentials into large-scale collagen simulations to compute reaction rates from predicted barriers, advancing mechanistic understanding of HAT and radical migration in peptides. We analyze scaling laws, model transferability, and cost-performance trade-offs, and outline strategies for improvement by combining ML potentials with transition state search algorithms and active learning. Our approach is generalizable to other biomolecular systems, enabling quantum-accurate simulations of chemical reactivity in complex environments.
Generating Highly Designable Proteins with Geometric Algebra Flow Matching
Nov 07, 2024



We introduce a generative model for protein backbone design utilizing geometric products and higher order message passing. In particular, we propose Clifford Frame Attention (CFA), an extension of the invariant point attention (IPA) architecture from AlphaFold2, in which the backbone residue frames and geometric features are represented in the projective geometric algebra. This enables to construct geometrically expressive messages between residues, including higher order terms, using the bilinear operations of the algebra. We evaluate our architecture by incorporating it into the framework of FrameFlow, a state-of-the-art flow matching model for protein backbone generation. The proposed model achieves high designability, diversity and novelty, while also sampling protein backbones that follow the statistical distribution of secondary structure elements found in naturally occurring proteins, a property so far only insufficiently achieved by many state-of-the-art generative models.
Grappa -- A Machine Learned Molecular Mechanics Force Field
Mar 25, 2024Simulating large molecular systems over long timescales requires force fields that are both accurate and efficient. In recent years, E(3) equivariant neural networks have lifted the tension between computational efficiency and accuracy of force fields, but they are still several orders of magnitude more expensive than classical molecular mechanics (MM) force fields. Here, we propose a novel machine learning architecture to predict MM parameters from the molecular graph, employing a graph attentional neural network and a transformer with symmetry-preserving positional encoding. The resulting force field, Grappa, outperforms established and other machine-learned MM force fields in terms of accuracy at the same computational efficiency and can be used in existing Molecular Dynamics (MD) engines like GROMACS and OpenMM. It predicts energies and forces of small molecules, peptides, RNA and - showcasing its extensibility to uncharted regions of chemical space - radicals at state-of-the-art MM accuracy. We demonstrate Grappa's transferability to macromolecules in MD simulations, during which large protein are kept stable and small proteins can fold. Our force field sets the stage for biomolecular simulations close to chemical accuracy, but with the same computational cost as established protein force fields.