 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLook the Other Way: Designing 'Positive' Molecules with Negative Data via Task Arithmetic
Jul 23, 2025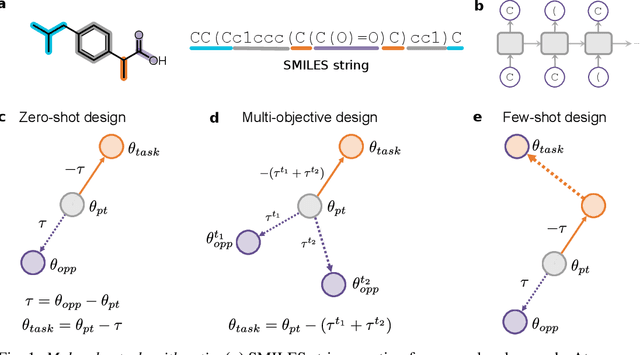
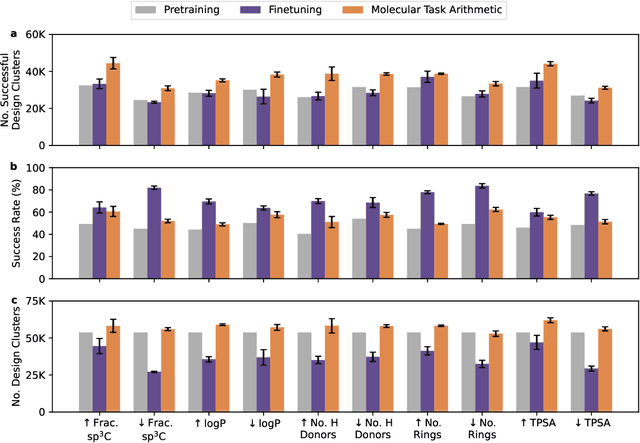
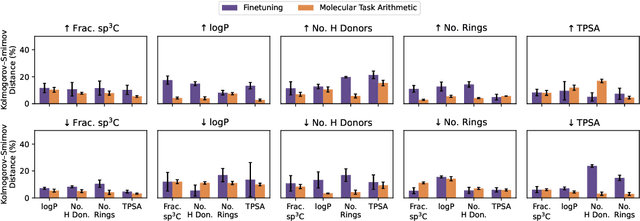
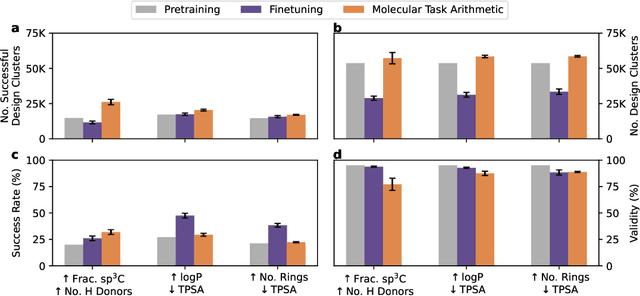
The scarcity of molecules with desirable properties (i.e., 'positive' molecules) is an inherent bottleneck for generative molecule design. To sidestep such obstacle, here we propose molecular task arithmetic: training a model on diverse and abundant negative examples to learn 'property directions' $--$ without accessing any positively labeled data $--$ and moving models in the opposite property directions to generate positive molecules. When analyzed on 20 zero-shot design experiments, molecular task arithmetic generated more diverse and successful designs than models trained on positive molecules. Moreover, we employed molecular task arithmetic in dual-objective and few-shot design tasks. We find that molecular task arithmetic can consistently increase the diversity of designs while maintaining desirable design properties. With its simplicity, data efficiency, and performance, molecular task arithmetic bears the potential to become the $\textit{de-facto}$ transfer learning strategy for de novo molecule design.
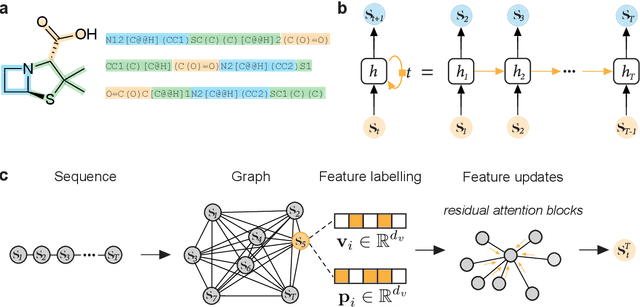
A Hitchhiker's Guide to Deep Chemical Language Processing for Bioactivity Prediction
Jul 16, 2024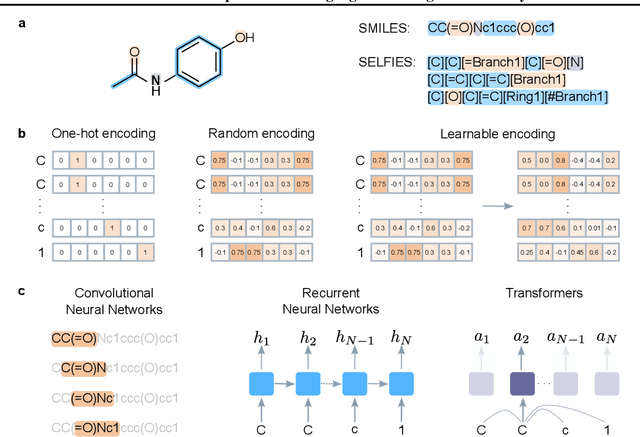
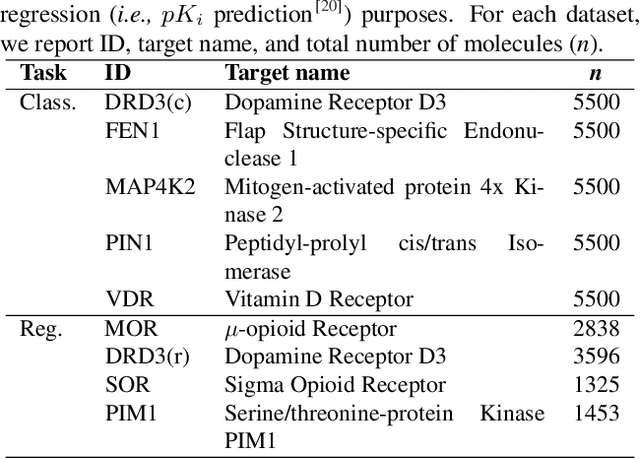

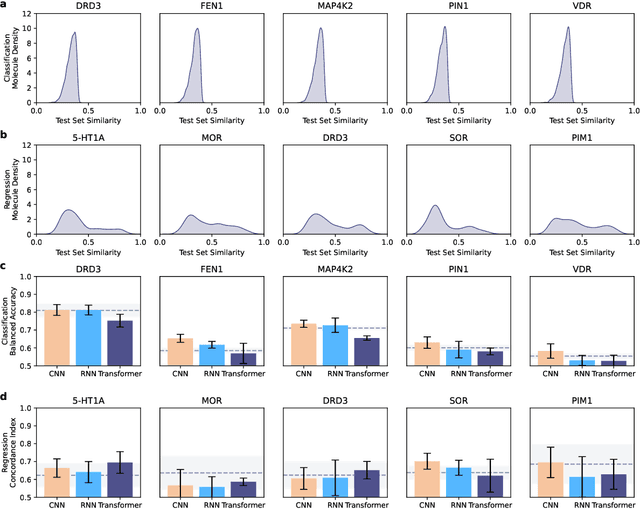
Deep learning has significantly accelerated drug discovery, with 'chemical language' processing (CLP) emerging as a prominent approach. CLP learns from molecular string representations (e.g., Simplified Molecular Input Line Entry Systems [SMILES] and Self-Referencing Embedded Strings [SELFIES]) with methods akin to natural language processing. Despite their growing importance, training predictive CLP models is far from trivial, as it involves many 'bells and whistles'. Here, we analyze the key elements of CLP training, to provide guidelines for newcomers and experts alike. Our study spans three neural network architectures, two string representations, three embedding strategies, across ten bioactivity datasets, for both classification and regression purposes. This 'hitchhiker's guide' not only underscores the importance of certain methodological choices, but it also equips researchers with practical recommendations on ideal choices, e.g., in terms of neural network architectures, molecular representations, and hyperparameter optimization.
Structure-based drug discovery with deep learning
Dec 26, 2022Artificial intelligence (AI) in the form of deep learning bears promise for drug discovery and chemical biology, $\textit{e.g.}$, to predict protein structure and molecular bioactivity, plan organic synthesis, and design molecules $\textit{de novo}$. While most of the deep learning efforts in drug discovery have focused on ligand-based approaches, structure-based drug discovery has the potential to tackle unsolved challenges, such as affinity prediction for unexplored protein targets, binding-mechanism elucidation, and the rationalization of related chemical kinetic properties. Advances in deep learning methodologies and the availability of accurate predictions for protein tertiary structure advocate for a $\textit{renaissance}$ in structure-based approaches for drug discovery guided by AI. This review summarizes the most prominent algorithmic concepts in structure-based deep learning for drug discovery, and forecasts opportunities, applications, and challenges ahead.
Geometric Deep Learning on Molecular Representations
Jul 30, 2021
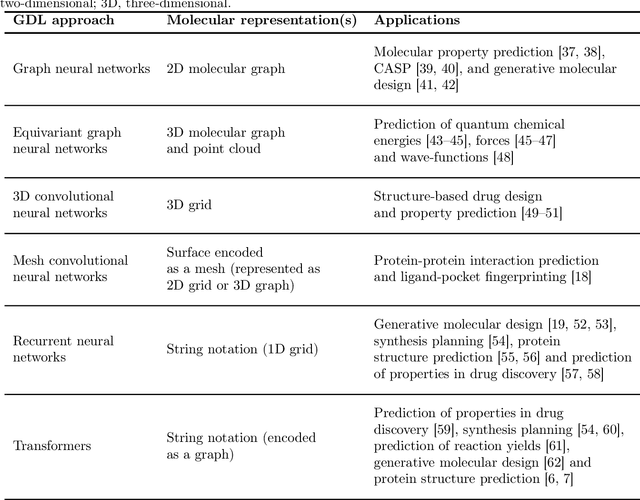
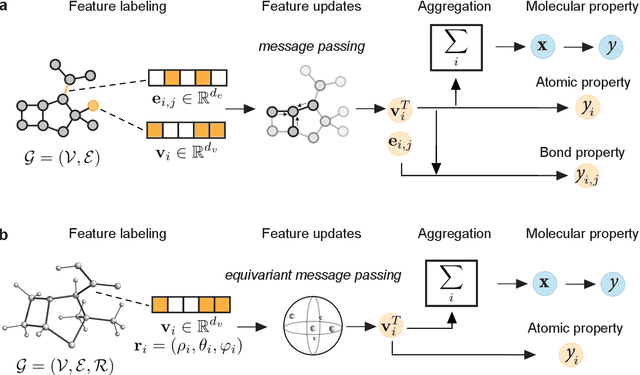
Geometric deep learning (GDL), which is based on neural network architectures that incorporate and process symmetry information, has emerged as a recent paradigm in artificial intelligence. GDL bears particular promise in molecular modeling applications, in which various molecular representations with different symmetry properties and levels of abstraction exist. This review provides a structured and harmonized overview of molecular GDL, highlighting its applications in drug discovery, chemical synthesis prediction, and quantum chemistry. Emphasis is placed on the relevance of the learned molecular features and their complementarity to well-established molecular descriptors. This review provides an overview of current challenges and opportunities, and presents a forecast of the future of GDL for molecular sciences.
Drug discovery with explainable artificial intelligence
Jul 02, 2020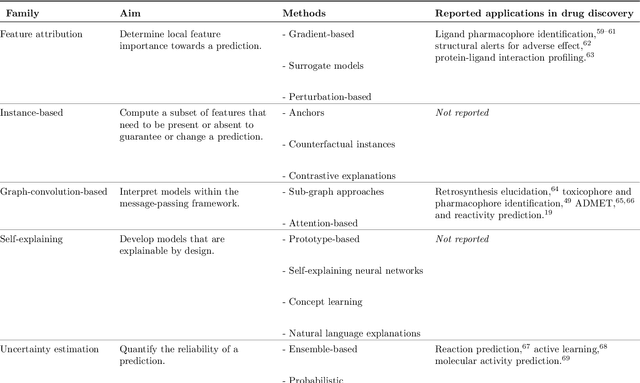
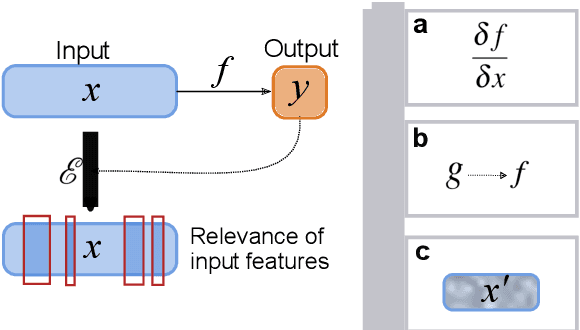
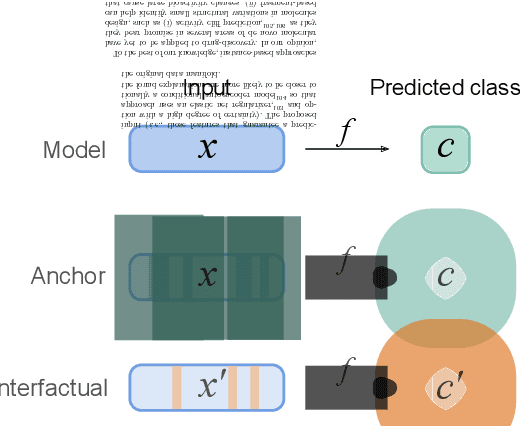
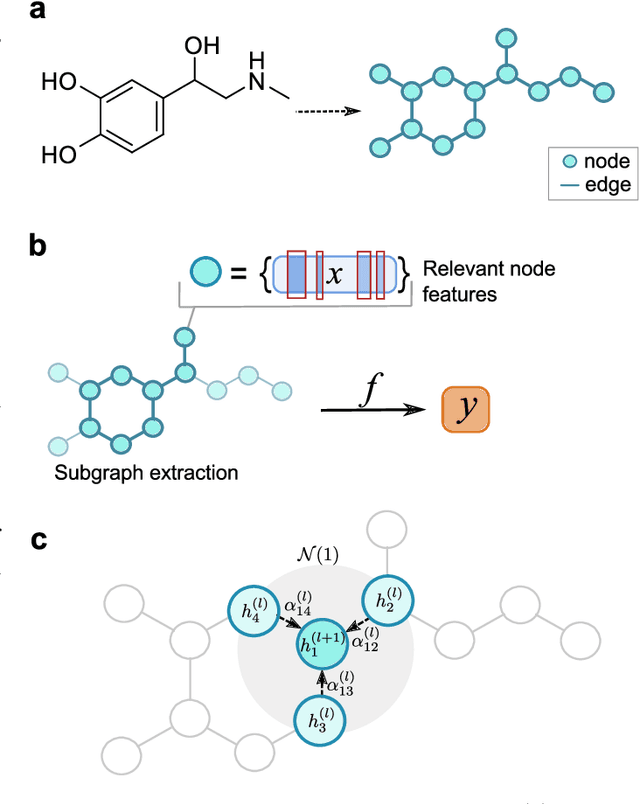
Deep learning bears promise for drug discovery, including advanced image analysis, prediction of molecular structure and function, and automated generation of innovative chemical entities with bespoke properties. Despite the growing number of successful prospective applications, the underlying mathematical models often remain elusive to interpretation by the human mind. There is a demand for 'explainable' deep learning methods to address the need for a new narrative of the machine language of the molecular sciences. This review summarizes the most prominent algorithmic concepts of explainable artificial intelligence, and dares a forecast of the future opportunities, potential applications, and remaining challenges.