 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Hitchhiker's Guide to Deep Chemical Language Processing for Bioactivity Prediction
Paper and Code
Jul 16, 2024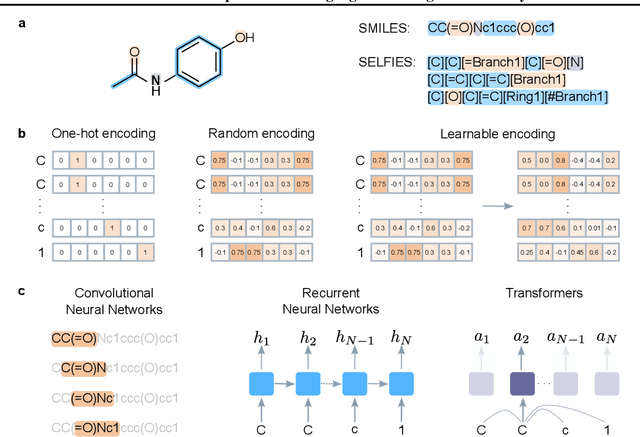
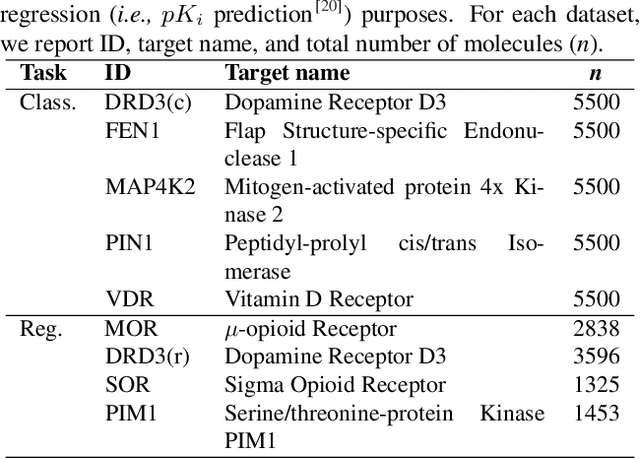

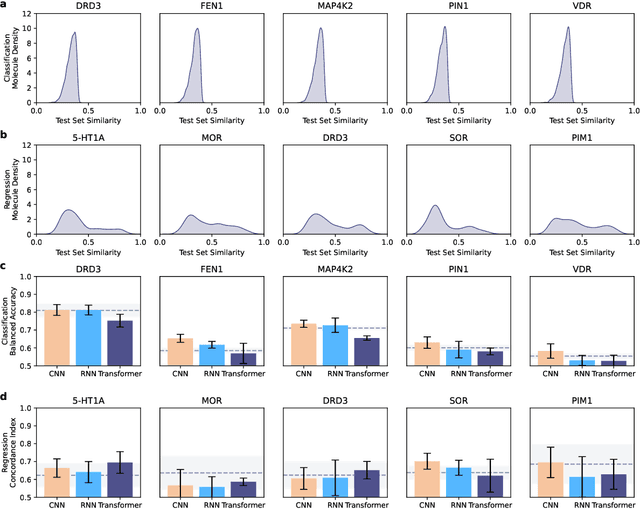
Deep learning has significantly accelerated drug discovery, with 'chemical language' processing (CLP) emerging as a prominent approach. CLP learns from molecular string representations (e.g., Simplified Molecular Input Line Entry Systems [SMILES] and Self-Referencing Embedded Strings [SELFIES]) with methods akin to natural language processing. Despite their growing importance, training predictive CLP models is far from trivial, as it involves many 'bells and whistles'. Here, we analyze the key elements of CLP training, to provide guidelines for newcomers and experts alike. Our study spans three neural network architectures, two string representations, three embedding strategies, across ten bioactivity datasets, for both classification and regression purposes. This 'hitchhiker's guide' not only underscores the importance of certain methodological choices, but it also equips researchers with practical recommendations on ideal choices, e.g., in terms of neural network architectures, molecular representations, and hyperparameter optimization.