 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDynamic Control of Explore/Exploit Trade-Off In Bayesian Optimization
Jul 03, 2018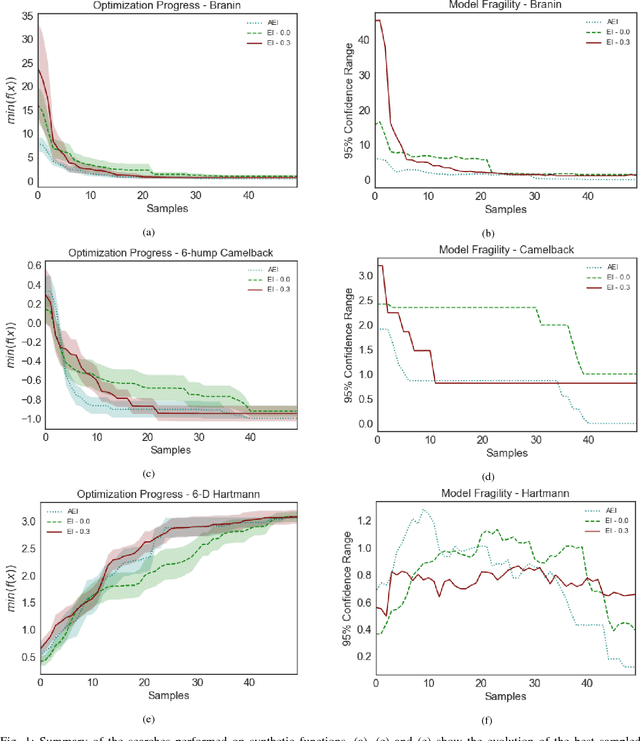
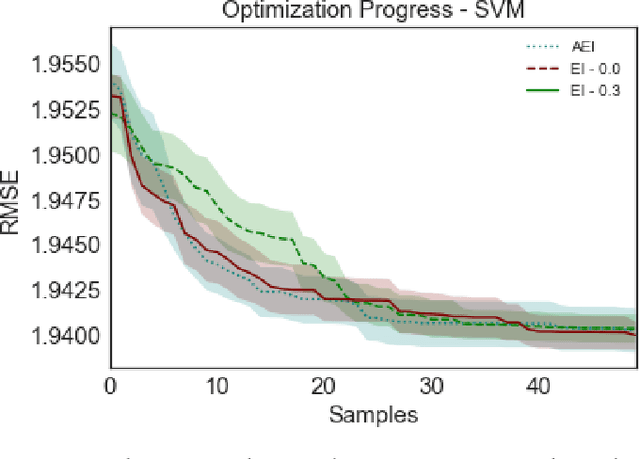
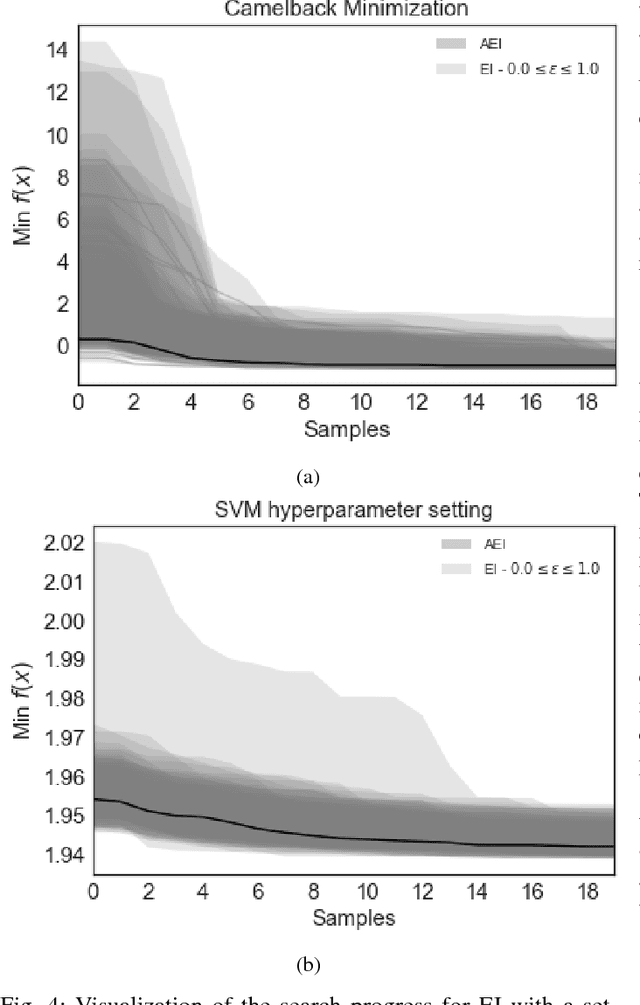
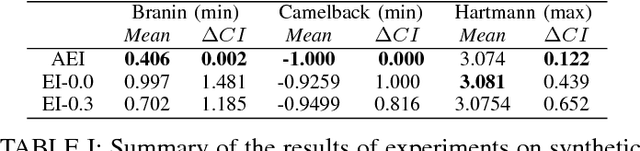
Bayesian optimization offers the possibility of optimizing black-box operations not accessible through traditional techniques. The success of Bayesian optimization methods such as Expected Improvement (EI) are significantly affected by the degree of trade-off between exploration and exploitation. Too much exploration can lead to inefficient optimization protocols, whilst too much exploitation leaves the protocol open to strong initial biases, and a high chance of getting stuck in a local minimum. Typically, a constant margin is used to control this trade-off, which results in yet another hyper-parameter to be optimized. We propose contextual improvement as a simple, yet effective heuristic to counter this - achieving a one-shot optimization strategy. Our proposed heuristic can be swiftly calculated and improves both the speed and robustness of discovery of optimal solutions. We demonstrate its effectiveness on both synthetic and real world problems and explore the unaccounted for uncertainty in the pre-determination of search hyperparameters controlling explore-exploit trade-off.
Space-Filling Curves as a Novel Crystal Structure Representation for Machine Learning Models
Aug 19, 2016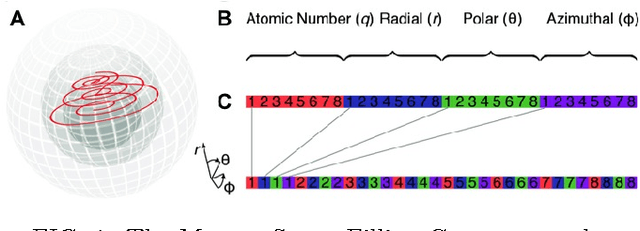
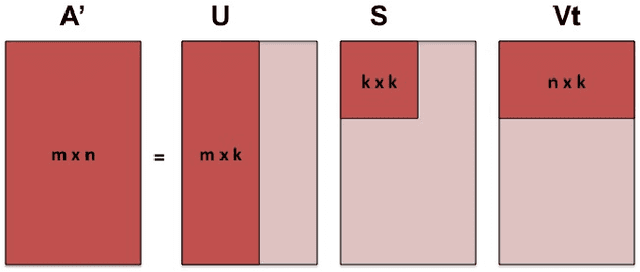
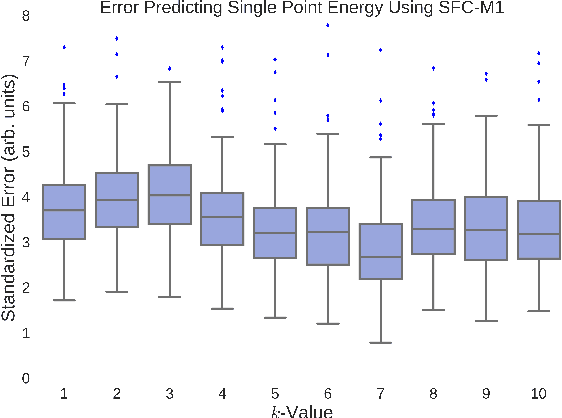
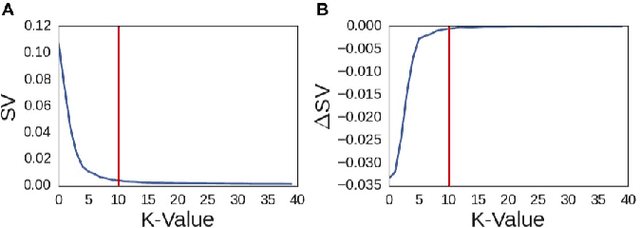
A fundamental problem in applying machine learning techniques for chemical problems is to find suitable representations for molecular and crystal structures. While the structure representations based on atom connectivities are prevalent for molecules, two-dimensional descriptors are not suitable for describing molecular crystals. In this work, we introduce the SFC-M family of feature representations, which are based on Morton space-filling curves, as an alternative means of representing crystal structures. Latent Semantic Indexing (LSI) was employed in a novel setting to reduce sparsity of feature representations. The quality of the SFC-M representations were assessed by using them in combination with artificial neural networks to predict Density Functional Theory (DFT) single point, Ewald summed, lattice, and many-body dispersion energies of 839 organic molecular crystal unit cells from the Cambridge Structural Database that consist of the elements C, H, N, and O. Promising initial results suggest that the SFC-M representations merit further exploration to improve its ability to predict solid-state properties of organic crystal structures