 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGROVER: Graph-guided Representation of Omics and Vision with Expert Regulation for Adaptive Spatial Multi-omics Fusion
Nov 13, 2025Effectively modeling multimodal spatial omics data is critical for understanding tissue complexity and underlying biological mechanisms. While spatial transcriptomics, proteomics, and epigenomics capture molecular features, they lack pathological morphological context. Integrating these omics with histopathological images is therefore essential for comprehensive disease tissue analysis. However, substantial heterogeneity across omics, imaging, and spatial modalities poses significant challenges. Naive fusion of semantically distinct sources often leads to ambiguous representations. Additionally, the resolution mismatch between high-resolution histology images and lower-resolution sequencing spots complicates spatial alignment. Biological perturbations during sample preparation further distort modality-specific signals, hindering accurate integration. To address these challenges, we propose Graph-guided Representation of Omics and Vision with Expert Regulation for Adaptive Spatial Multi-omics Fusion (GROVER), a novel framework for adaptive integration of spatial multi-omics data. GROVER leverages a Graph Convolutional Network encoder based on Kolmogorov-Arnold Networks to capture the nonlinear dependencies between each modality and its associated spatial structure, thereby producing expressive, modality-specific embeddings. To align these representations, we introduce a spot-feature-pair contrastive learning strategy that explicitly optimizes the correspondence across modalities at each spot. Furthermore, we design a dynamic expert routing mechanism that adaptively selects informative modalities for each spot while suppressing noisy or low-quality inputs. Experiments on real-world spatial omics datasets demonstrate that GROVER outperforms state-of-the-art baselines, providing a robust and reliable solution for multimodal integration.
scFusionTTT: Single-cell transcriptomics and proteomics fusion with Test-Time Training layers
Oct 17, 2024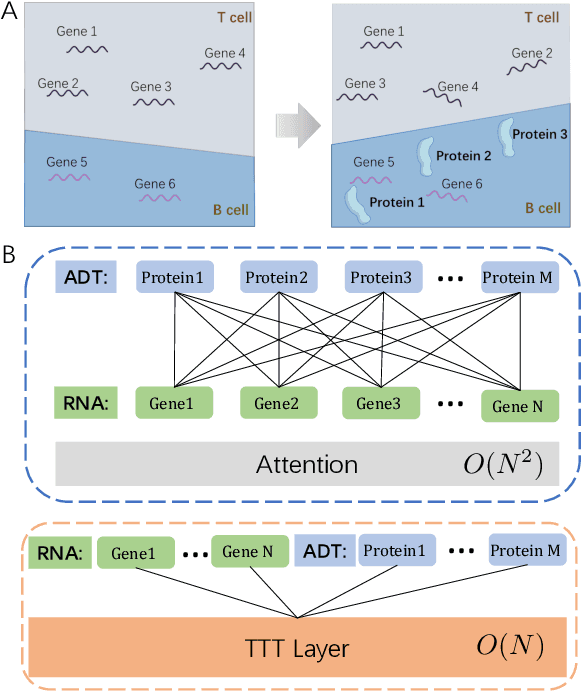
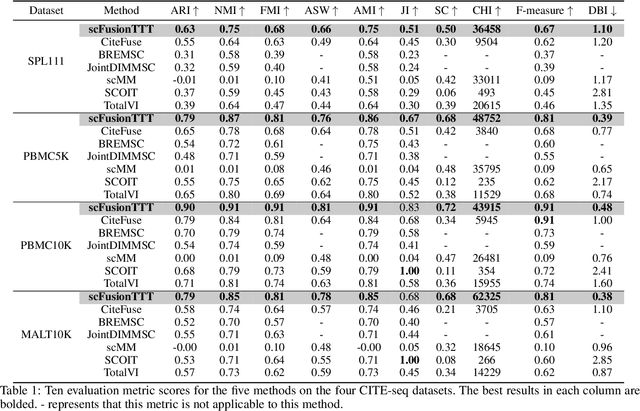
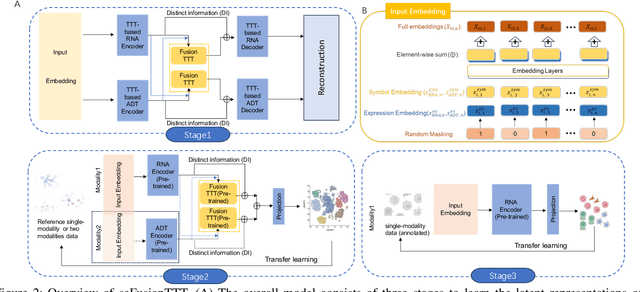
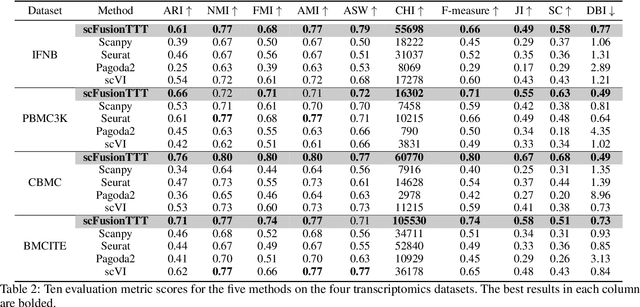
Single-cell multi-omics (scMulti-omics) refers to the paired multimodal data, such as Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq), where the regulation of each cell was measured from different modalities, i.e. genes and proteins. scMulti-omics can reveal heterogeneity inside tumors and understand the distinct genetic properties of diverse cell types, which is crucial to targeted therapy. Currently, deep learning methods based on attention structures in the bioinformatics area face two challenges. The first challenge is the vast number of genes in a single cell. Traditional attention-based modules struggled to effectively leverage all gene information due to their limited capacity for long-context learning and high-complexity computing. The second challenge is that genes in the human genome are ordered and influence each other's expression. Most of the methods ignored this sequential information. The recently introduced Test-Time Training (TTT) layer is a novel sequence modeling approach, particularly suitable for handling long contexts like genomics data because TTT layer is a linear complexity sequence modeling structure and is better suited to data with sequential relationships. In this paper, we propose scFusionTTT, a novel method for Single-Cell multimodal omics Fusion with TTT-based masked autoencoder. Of note, we combine the order information of genes and proteins in the human genome with the TTT layer, fuse multimodal omics, and enhance unimodal omics analysis. Finally, the model employs a three-stage training strategy, which yielded the best performance across most metrics in four multimodal omics datasets and four unimodal omics datasets, demonstrating the superior performance of our model. The dataset and code will be available on https://github.com/DM0815/scFusionTTT.