 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Opioid Use Disorder Risk Modelling through Behavioral and Genetic Feature Integration
Sep 19, 2023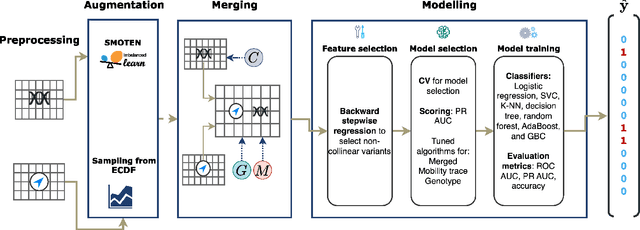
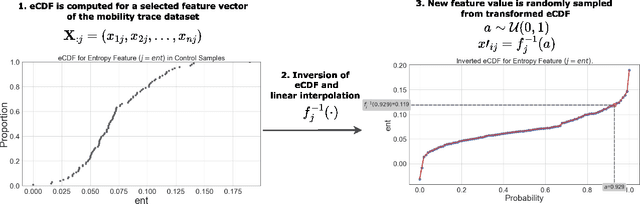
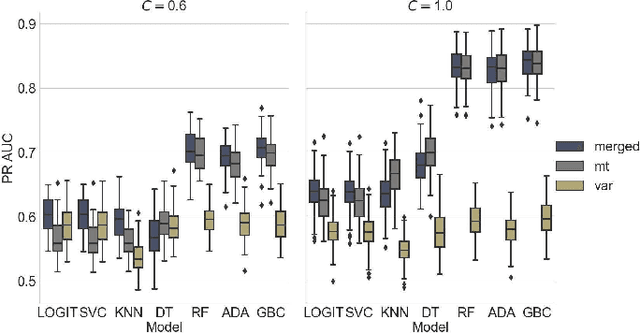
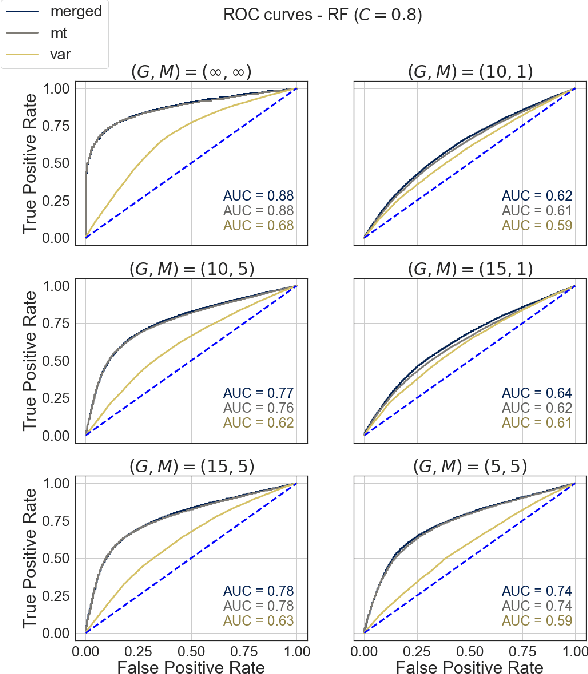
Opioids are an effective analgesic for acute and chronic pain, but also carry a considerable risk of addiction leading to millions of opioid use disorder (OUD) cases and tens of thousands of premature deaths in the United States yearly. Estimating OUD risk prior to prescription could improve the efficacy of treatment regimens, monitoring programs, and intervention strategies, but risk estimation is typically based on self-reported data or questionnaires. We develop an experimental design and computational methods that combines genetic variants associated with OUD with behavioral features extracted from GPS and Wi-Fi spatiotemporal coordinates to assess OUD risk. Since both OUD mobility and genetic data do not exist for the same cohort, we develop algorithms to (1) generate mobility features from empirical distributions and (2) synthesize mobility and genetic samples assuming a level of comorbidity and relative risks. We show that integrating genetic and mobility modalities improves risk modelling using classification accuracy, area under the precision-recall and receiver operator characteristic curves, and $F_1$ score. Interpreting the fitted models suggests that mobility features have more influence on OUD risk, although the genetic contribution was significant, particularly in linear models. While there exists concerns with respect to privacy, security, bias, and generalizability that must be evaluated in clinical trials before being implemented in practice, our framework provides preliminary evidence that behavioral and genetic features may improve OUD risk estimation to assist with personalized clinical decision-making.
Bayesian Reconstruction and Differential Testing of Excised mRNA
Nov 14, 2022



Characterizing the differential excision of mRNA is critical for understanding the functional complexity of a cell or tissue, from normal developmental processes to disease pathogenesis. Most transcript reconstruction methods infer full-length transcripts from high-throughput sequencing data. However, this is a challenging task due to incomplete annotations and the differential expression of transcripts across cell-types, tissues, and experimental conditions. Several recent methods circumvent these difficulties by considering local splicing events, but these methods lose transcript-level splicing information and may conflate transcripts. We develop the first probabilistic model that reconciles the transcript and local splicing perspectives. First, we formalize the sequence of mRNA excisions (SME) reconstruction problem, which aims to assemble variable-length sequences of mRNA excisions from RNA-sequencing data. We then present a novel hierarchical Bayesian admixture model for the Reconstruction of Excised mRNA (BREM). BREM interpolates between local splicing events and full-length transcripts and thus focuses only on SMEs that have high posterior probability. We develop posterior inference algorithms based on Gibbs sampling and local search of independent sets and characterize differential SME usage using generalized linear models based on converged BREM model parameters. We show that BREM achieves higher F1 score for reconstruction tasks and improved accuracy and sensitivity in differential splicing when compared with four state-of-the-art transcript and local splicing methods on simulated data. Lastly, we evaluate BREM on both bulk and scRNA sequencing data based on transcript reconstruction, novelty of transcripts produced, model sensitivity to hyperparameters, and a functional analysis of differentially expressed SMEs, demonstrating that BREM captures relevant biological signal.