 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Bigger the Better? Accurate Molecular Potential Energy Surfaces from Minimalist Neural Networks
Nov 27, 2024



Atomistic simulations are a powerful tool for studying the dynamics of molecules, proteins, and materials on wide time and length scales. Their reliability and predictiveness, however, depend directly on the accuracy of the underlying potential energy surface (PES). Guided by the principle of parsimony this work introduces KerNN, a combined kernel/neural network-based approach to represent molecular PESs. Compared to state-of-the-art neural network PESs the number of learnable parameters of KerNN is significantly reduced. This speeds up training and evaluation times by several orders of magnitude while retaining high prediction accuracy. Importantly, using kernels as the features also improves the extrapolation capabilities of KerNN far beyond the coverage provided by the training data which solves a general problem of NN-based PESs. KerNN applied to spectroscopy and reaction dynamics shows excellent performance on test set statistics and observables including vibrational bands computed from classical and quantum simulations.
Machine Learning Product State Distributions from Initial Reactant States for a Reactive Atom-Diatom Collision System
Nov 05, 2021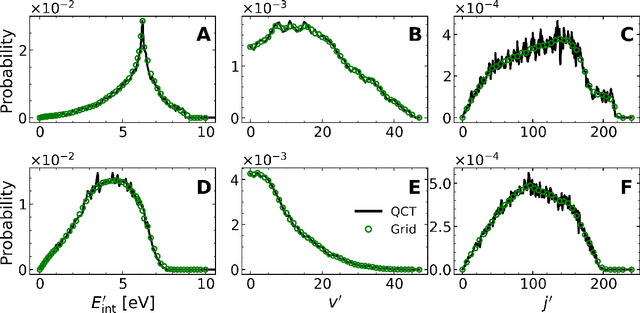
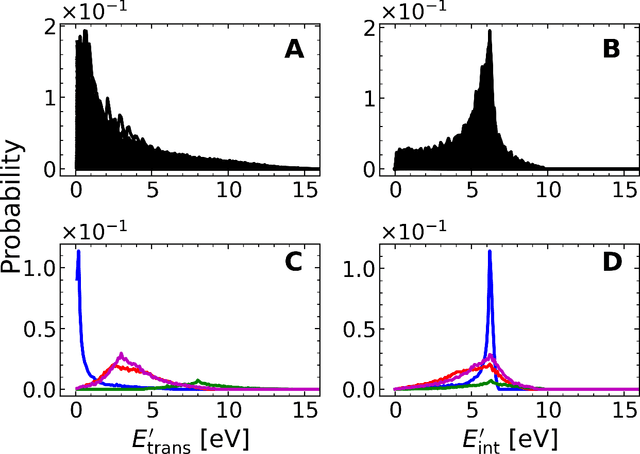
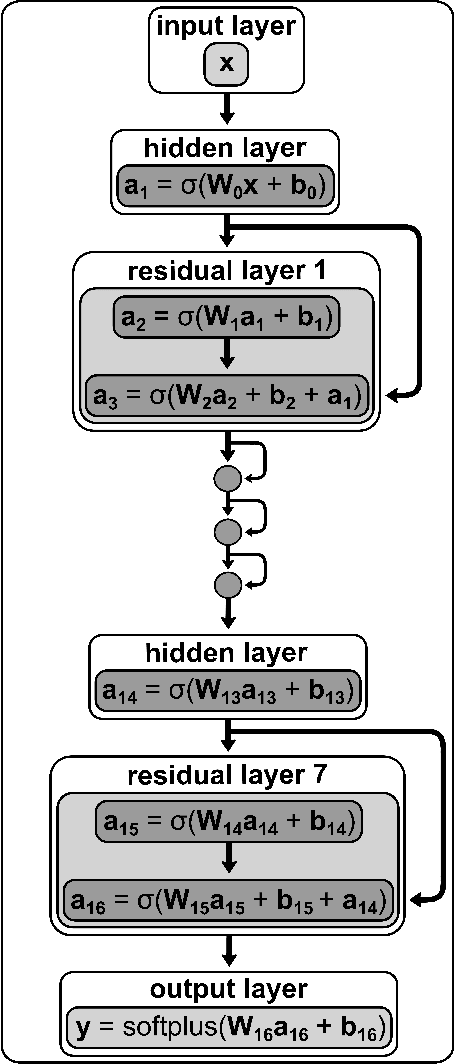
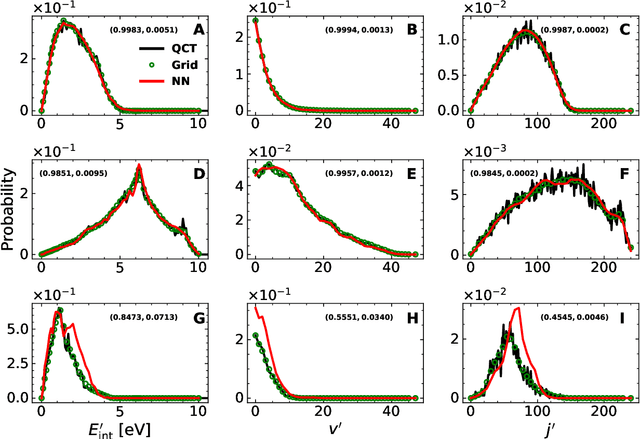
A machine learned (ML) model for predicting product state distributions from specific initial states (state-to-distribution or STD) for reactive atom-diatom collisions is presented and quantitatively tested for the N($^4$S)+O$_{2}$(X$^3 \Sigma_{\rm g}^{-}$) $\rightarrow$ NO(X$^2\Pi$) +O($^3$P) reaction. The reference data set for training the neural network (NN) consists of final state distributions determined from explicit quasi-classical trajectory (QCT) simulations for $\sim 2000$ initial conditions. Overall, the prediction accuracy as quantified by the root-mean-squared difference $(\sim 0.003)$ and the $R^2$ $(\sim 0.99)$ between the reference QCT and predictions of the STD model is high for the test set and off-grid state specific initial conditions and for initial conditions drawn from reactant state distributions characterized by translational, rotational and vibrational temperatures. Compared with a more coarse grained distribution-to-distribution (DTD) model evaluated on the same initial state distributions, the STD model shows comparable performance with the additional benefit of the state resolution in the reactant preparation. Starting from specific initial states also leads to a more diverse range of final state distributions which requires a more expressive neural network to be used compared with DTD. Direct comparison between explicit QCT simulations, the STD model, and the widely used Larsen-Borgnakke (LB) model shows that the STD model is quantitative whereas the LB model is qualitative at best for rotational distributions $P(j')$ and fails for vibrational distributions $P(v')$. As such the STD model can be well-suited for simulating nonequilibrium high-speed flows, e.g., using the direct simulation Monte Carlo method.