 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlowMol3: Flow Matching for 3D De Novo Small-Molecule Generation
Aug 18, 2025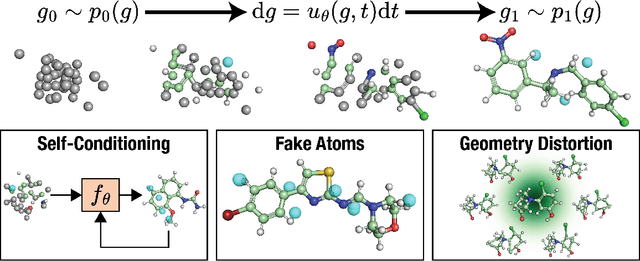
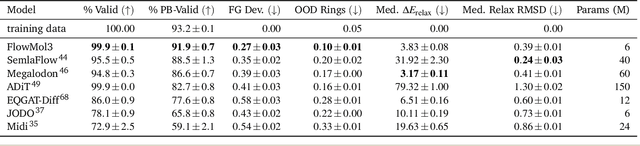
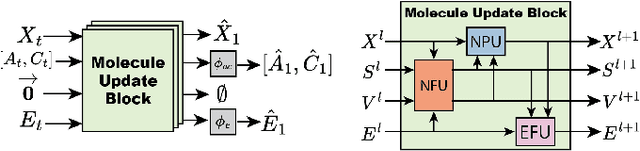
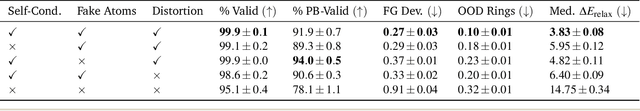
A generative model capable of sampling realistic molecules with desired properties could accelerate chemical discovery across a wide range of applications. Toward this goal, significant effort has focused on developing models that jointly sample molecular topology and 3D structure. We present FlowMol3, an open-source, multi-modal flow matching model that advances the state of the art for all-atom, small-molecule generation. Its substantial performance gains over previous FlowMol versions are achieved without changes to the graph neural network architecture or the underlying flow matching formulation. Instead, FlowMol3's improvements arise from three architecture-agnostic techniques that incur negligible computational cost: self-conditioning, fake atoms, and train-time geometry distortion. FlowMol3 achieves nearly 100% molecular validity for drug-like molecules with explicit hydrogens, more accurately reproduces the functional group composition and geometry of its training data, and does so with an order of magnitude fewer learnable parameters than comparable methods. We hypothesize that these techniques mitigate a general pathology affecting transport-based generative models, enabling detection and correction of distribution drift during inference. Our results highlight simple, transferable strategies for improving the stability and quality of diffusion- and flow-based molecular generative models.
Exploring Discrete Flow Matching for 3D De Novo Molecule Generation
Nov 25, 2024Deep generative models that produce novel molecular structures have the potential to facilitate chemical discovery. Flow matching is a recently proposed generative modeling framework that has achieved impressive performance on a variety of tasks including those on biomolecular structures. The seminal flow matching framework was developed only for continuous data. However, de novo molecular design tasks require generating discrete data such as atomic elements or sequences of amino acid residues. Several discrete flow matching methods have been proposed recently to address this gap. In this work we benchmark the performance of existing discrete flow matching methods for 3D de novo small molecule generation and provide explanations of their differing behavior. As a result we present FlowMol-CTMC, an open-source model that achieves state of the art performance for 3D de novo design with fewer learnable parameters than existing methods. Additionally, we propose the use of metrics that capture molecule quality beyond local chemical valency constraints and towards higher-order structural motifs. These metrics show that even though basic constraints are satisfied, the models tend to produce unusual and potentially problematic functional groups outside of the training data distribution. Code and trained models for reproducing this work are available at \url{https://github.com/dunni3/FlowMol}.
SPRINT Enables Interpretable and Ultra-Fast Virtual Screening against Thousands of Proteomes
Nov 23, 2024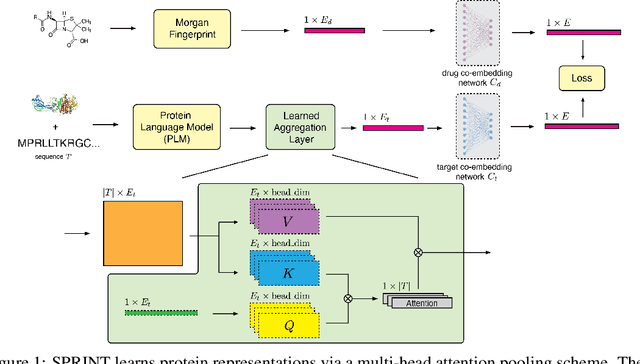
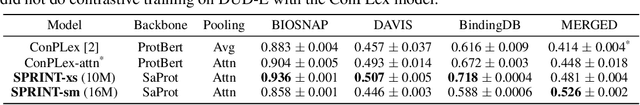

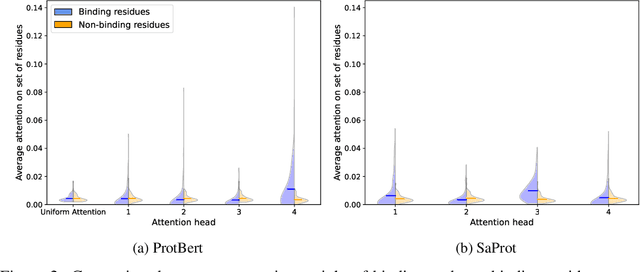
Virtual screening of small molecules against protein targets can accelerate drug discovery and development by predicting drug-target interactions (DTIs). However, structure-based methods like molecular docking are too slow to allow for broad proteome-scale screens, limiting their application in screening for off-target effects or new molecular mechanisms. Recently, vector-based methods using protein language models (PLMs) have emerged as a complementary approach that bypasses explicit 3D structure modeling. Here, we develop SPRINT, a vector-based approach for screening entire chemical libraries against whole proteomes for DTIs and novel mechanisms of action. SPRINT improves on prior work by using a self-attention based architecture and structure-aware PLMs to learn drug-target co-embeddings for binder prediction, search, and retrieval. SPRINT achieves SOTA enrichment factors in virtual screening on LIT-PCBA and DTI classification benchmarks, while providing interpretability in the form of residue-level attention maps. In addition to being both accurate and interpretable, SPRINT is ultra-fast: querying the whole human proteome against the ENAMINE Real Database (6.7B drugs) for the 100 most likely binders per protein takes 16 minutes. SPRINT promises to enable virtual screening at an unprecedented scale, opening up new opportunities for in silico drug repurposing and development. SPRINT is available on the web as ColabScreen: https://bit.ly/colab-screen