 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn artificial intelligence framework for end-to-end rare disease phenotyping from clinical notes using large language models
Feb 23, 2026Phenotyping is fundamental to rare disease diagnosis, but manual curation of structured phenotypes from clinical notes is labor-intensive and difficult to scale. Existing artificial intelligence approaches typically optimize individual components of phenotyping but do not operationalize the full clinical workflow of extracting features from clinical text, standardizing them to Human Phenotype Ontology (HPO) terms, and prioritizing diagnostically informative HPO terms. We developed RARE-PHENIX, an end-to-end AI framework for rare disease phenotyping that integrates large language model-based phenotype extraction, ontology-grounded standardization to HPO terms, and supervised ranking of diagnostically informative phenotypes. We trained RARE-PHENIX using data from 2,671 patients across 11 Undiagnosed Diseases Network clinical sites, and externally validated it on 16,357 real-world clinical notes from Vanderbilt University Medical Center. Using clinician-curated HPO terms as the gold standard, RARE-PHENIX consistently outperformed a state-of-the-art deep learning baseline (PhenoBERT) across ontology-based similarity and precision-recall-F1 metrics in end-to-end evaluation (i.e., ontology-based similarity of 0.70 vs. 0.58). Ablation analyses demonstrated performance improvements with the addition of each module in RARE-PHENIX (extraction, standardization, and prioritization), supporting the value of modeling the full clinical phenotyping workflow. By modeling phenotyping as a clinically aligned workflow rather than a single extraction task, RARE-PHENIX provides structured, ranked phenotypes that are more concordant with clinician curation and has the potential to support human-in-the-loop rare disease diagnosis in real-world settings.
Identifying and Extracting Rare Disease Phenotypes with Large Language Models
Jun 22, 2023Rare diseases (RDs) are collectively common and affect 300 million people worldwide. Accurate phenotyping is critical for informing diagnosis and treatment, but RD phenotypes are often embedded in unstructured text and time-consuming to extract manually. While natural language processing (NLP) models can perform named entity recognition (NER) to automate extraction, a major bottleneck is the development of a large, annotated corpus for model training. Recently, prompt learning emerged as an NLP paradigm that can lead to more generalizable results without any (zero-shot) or few labeled samples (few-shot). Despite growing interest in ChatGPT, a revolutionary large language model capable of following complex human prompts and generating high-quality responses, none have studied its NER performance for RDs in the zero- and few-shot settings. To this end, we engineered novel prompts aimed at extracting RD phenotypes and, to the best of our knowledge, are the first the establish a benchmark for evaluating ChatGPT's performance in these settings. We compared its performance to the traditional fine-tuning approach and conducted an in-depth error analysis. Overall, fine-tuning BioClinicalBERT resulted in higher performance (F1 of 0.689) than ChatGPT (F1 of 0.472 and 0.591 in the zero- and few-shot settings, respectively). Despite this, ChatGPT achieved similar or higher accuracy for certain entities (i.e., rare diseases and signs) in the one-shot setting (F1 of 0.776 and 0.725). This suggests that with appropriate prompt engineering, ChatGPT has the potential to match or outperform fine-tuned language models for certain entity types with just one labeled sample. While the proliferation of large language models may provide opportunities for supporting RD diagnosis and treatment, researchers and clinicians should critically evaluate model outputs and be well-informed of their limitations.
Multi-study R-learner for Heterogeneous Treatment Effect Estimation
Jun 16, 2023Estimating heterogeneous treatment effects is crucial for informing personalized treatment strategies and policies. While multiple studies can improve the accuracy and generalizability of results, leveraging them for estimation is statistically challenging. Existing approaches often assume identical heterogeneous treatment effects across studies, but this may be violated due to various sources of between-study heterogeneity, including differences in study design, confounders, and sample characteristics. To this end, we propose a unifying framework for multi-study heterogeneous treatment effect estimation that is robust to between-study heterogeneity in the nuisance functions and treatment effects. Our approach, the multi-study R-learner, extends the R-learner to obtain principled statistical estimation with modern machine learning (ML) in the multi-study setting. The multi-study R-learner is easy to implement and flexible in its ability to incorporate ML for estimating heterogeneous treatment effects, nuisance functions, and membership probabilities, which borrow strength across heterogeneous studies. It achieves robustness in confounding adjustment through its loss function and can leverage both randomized controlled trials and observational studies. We provide asymptotic guarantees for the proposed method in the case of series estimation and illustrate using real cancer data that it has the lowest estimation error compared to existing approaches in the presence of between-study heterogeneity.
Multi-Study Boosting: Theoretical Considerations for Merging vs. Ensembling
Jul 13, 2022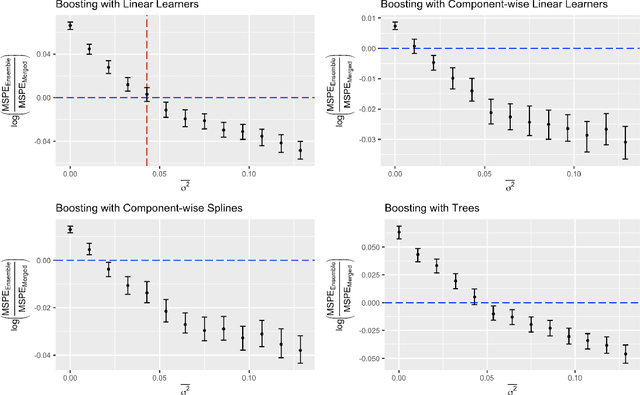
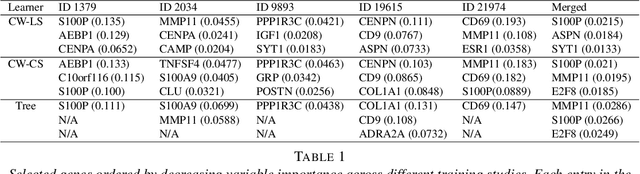
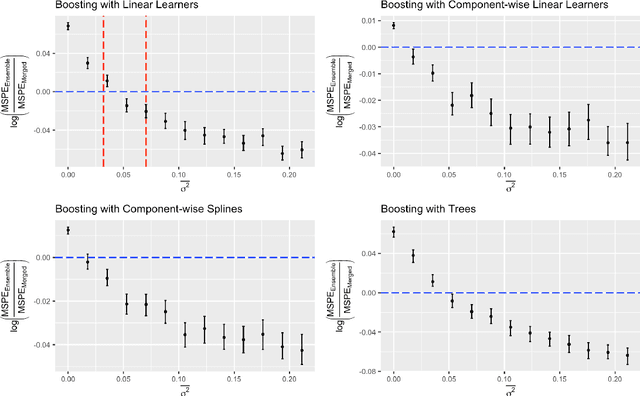
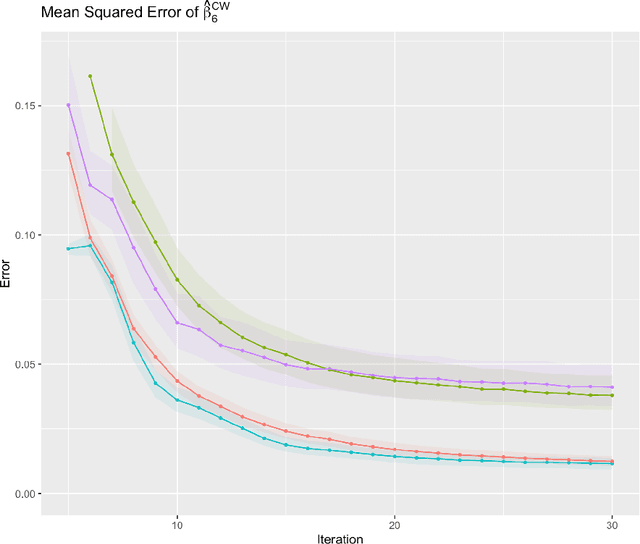
Cross-study replicability is a powerful model evaluation criterion that emphasizes generalizability of predictions. When training cross-study replicable prediction models, it is critical to decide between merging and treating the studies separately. We study boosting algorithms in the presence of potential heterogeneity in predictor-outcome relationships across studies and compare two multi-study learning strategies: 1) merging all the studies and training a single model, and 2) multi-study ensembling, which involves training a separate model on each study and ensembling the resulting predictions. In the regression setting, we provide theoretical guidelines based on an analytical transition point to determine whether it is more beneficial to merge or to ensemble for boosting with linear learners. In addition, we characterize a bias-variance decomposition of estimation error for boosting with component-wise linear learners. We verify the theoretical transition point result in simulation and illustrate how it can guide the decision on merging vs. ensembling in an application to breast cancer gene expression data.