 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLitGen: Genetic Literature Recommendation Guided by Human Explanations
Sep 24, 2019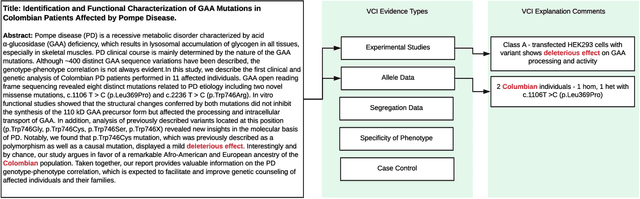
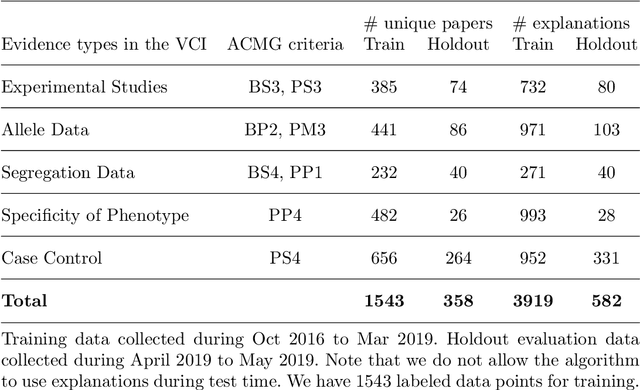
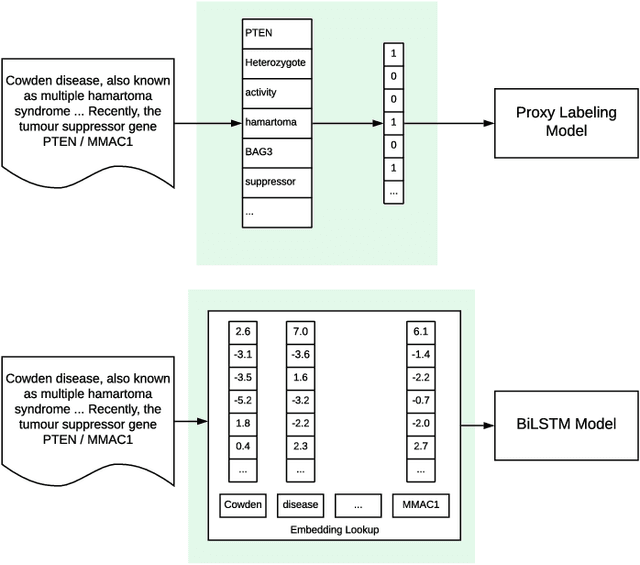
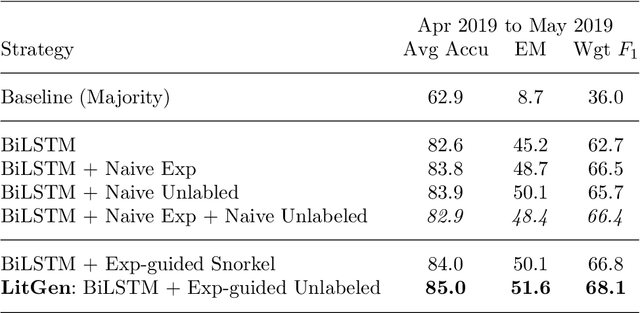
As genetic sequencing costs decrease, the lack of clinical interpretation of variants has become the bottleneck in using genetics data. A major rate limiting step in clinical interpretation is the manual curation of evidence in the genetic literature by highly trained biocurators. What makes curation particularly time-consuming is that the curator needs to identify papers that study variant pathogenicity using different types of approaches and evidences---e.g. biochemical assays or case control analysis. In collaboration with the Clinical Genomic Resource (ClinGen)---the flagship NIH program for clinical curation---we propose the first machine learning system, LitGen, that can retrieve papers for a particular variant and filter them by specific evidence types used by curators to assess for pathogenicity. LitGen uses semi-supervised deep learning to predict the type of evidence provided by each paper. It is trained on papers annotated by ClinGen curators and systematically evaluated on new test data collected by ClinGen. LitGen further leverages rich human explanations and unlabeled data to gain 7.9%-12.6% relative performance improvement over models learned only on the annotated papers. It is a useful framework to improve clinical variant curation.
DeepTag: inferring all-cause diagnoses from clinical notes in under-resourced medical domain
Sep 03, 2018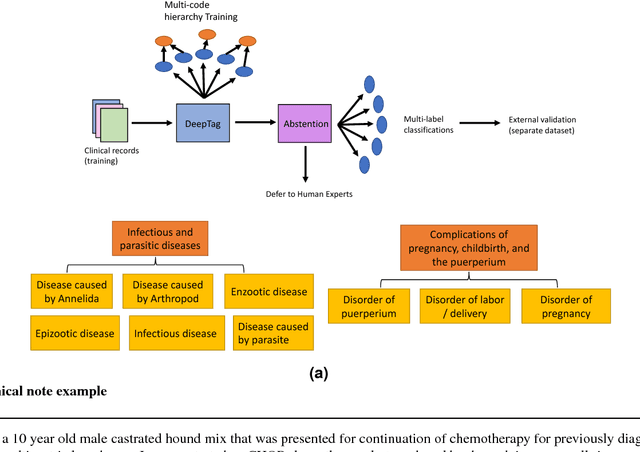
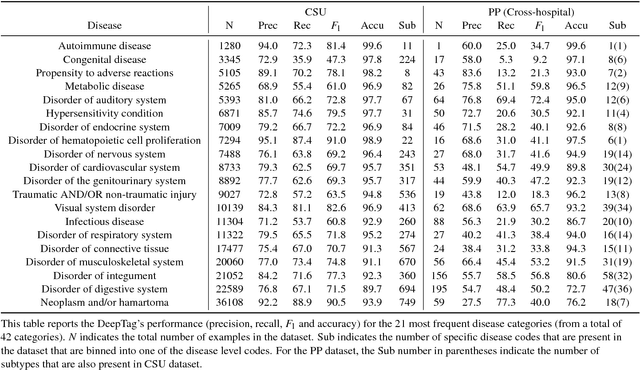
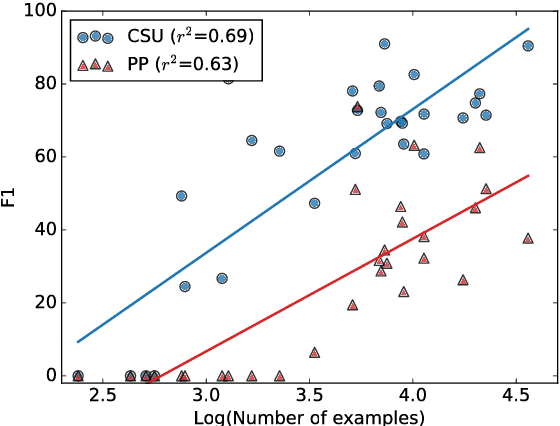
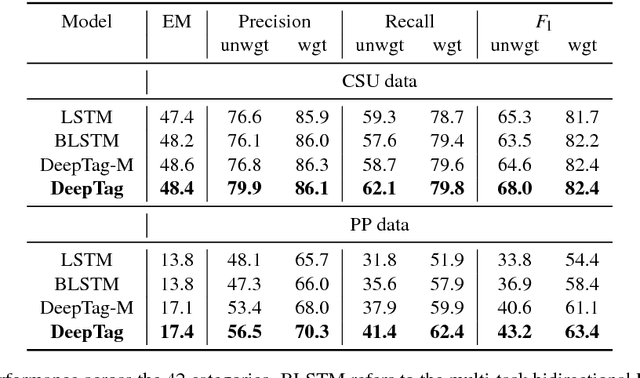
Large scale veterinary clinical records can become a powerful resource for patient care and research. However, clinicians lack the time and resource to annotate patient records with standard medical diagnostic codes and most veterinary visits are captured in free text notes. The lack of standard coding makes it challenging to use the clinical data to improve patient care. It is also a major impediment to cross-species translational research, which relies on the ability to accurately identify patient cohorts with specific diagnostic criteria in humans and animals. In order to reduce the coding burden for veterinary clinical practice and aid translational research, we have developed a deep learning algorithm, DeepTag, which automatically infers diagnostic codes from veterinary free text notes. DeepTag is trained on a newly curated dataset of 112,558 veterinary notes manually annotated by experts. DeepTag extends multi-task LSTM with an improved hierarchical objective that captures the semantic structures between diseases. To foster human-machine collaboration, DeepTag also learns to abstain in examples when it is uncertain and defers them to human experts, resulting in improved performance. DeepTag accurately infers disease codes from free text even in challenging cross-hospital settings where the text comes from different clinical settings than the ones used for training. It enables automated disease annotation across a broad range of clinical diagnoses with minimal pre-processing. The technical framework in this work can be applied in other medical domains that currently lack medical coding resources.
Network Enhancement: a general method to denoise weighted biological networks
Jun 01, 2018



Networks are ubiquitous in biology where they encode connectivity patterns at all scales of organization, from molecular to the biome. However, biological networks are noisy due to the limitations of measurement technology and inherent natural variation, which can hamper discovery of network patterns and dynamics. We propose Network Enhancement (NE), a method for improving the signal-to-noise ratio of undirected, weighted networks. NE uses a doubly stochastic matrix operator that induces sparsity and provides a closed-form solution that increases spectral eigengap of the input network. As a result, NE removes weak edges, enhances real connections, and leads to better downstream performance. Experiments show that NE improves gene function prediction by denoising tissue-specific interaction networks, alleviates interpretation of noisy Hi-C contact maps from the human genome, and boosts fine-grained identification accuracy of species. Our results indicate that NE is widely applicable for denoising biological networks.