 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLitGen: Genetic Literature Recommendation Guided by Human Explanations
Sep 24, 2019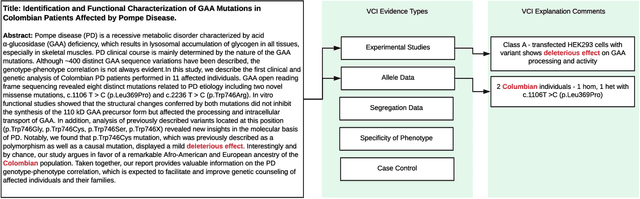
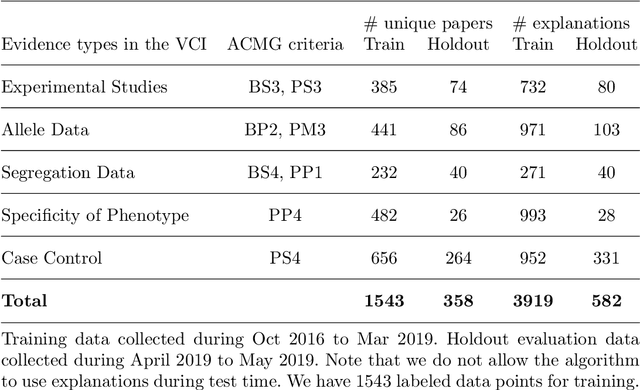
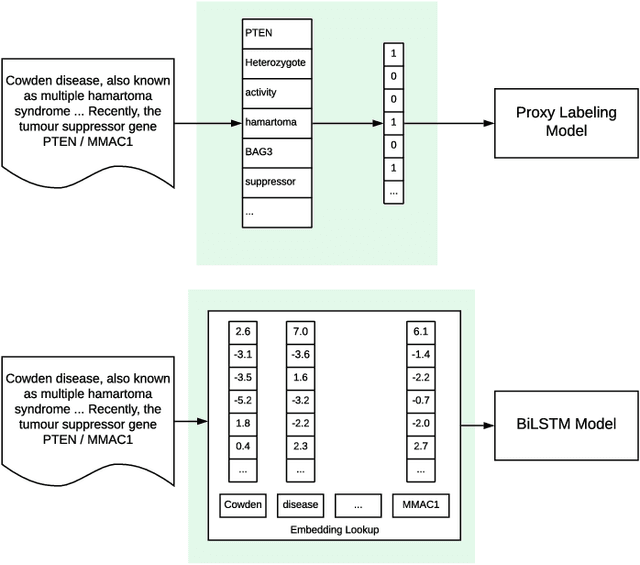
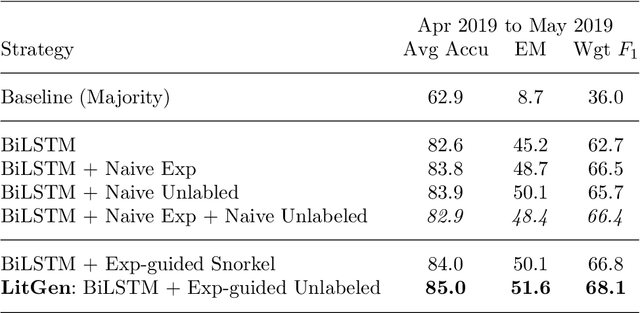
As genetic sequencing costs decrease, the lack of clinical interpretation of variants has become the bottleneck in using genetics data. A major rate limiting step in clinical interpretation is the manual curation of evidence in the genetic literature by highly trained biocurators. What makes curation particularly time-consuming is that the curator needs to identify papers that study variant pathogenicity using different types of approaches and evidences---e.g. biochemical assays or case control analysis. In collaboration with the Clinical Genomic Resource (ClinGen)---the flagship NIH program for clinical curation---we propose the first machine learning system, LitGen, that can retrieve papers for a particular variant and filter them by specific evidence types used by curators to assess for pathogenicity. LitGen uses semi-supervised deep learning to predict the type of evidence provided by each paper. It is trained on papers annotated by ClinGen curators and systematically evaluated on new test data collected by ClinGen. LitGen further leverages rich human explanations and unlabeled data to gain 7.9%-12.6% relative performance improvement over models learned only on the annotated papers. It is a useful framework to improve clinical variant curation.