 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024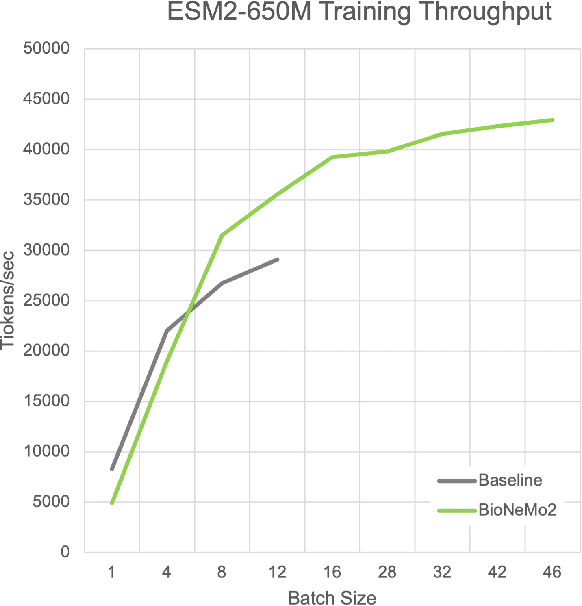
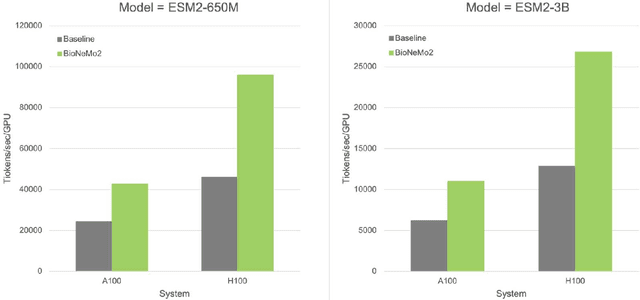
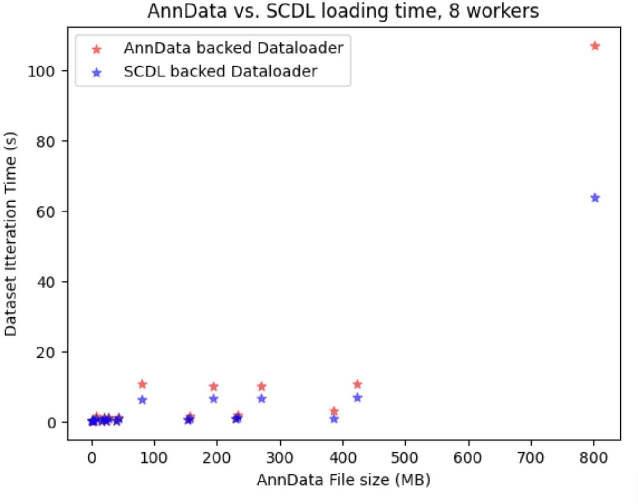
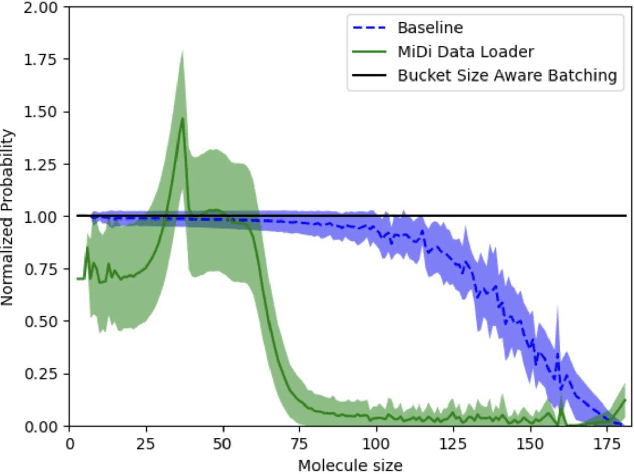
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
Improving Inverse Folding for Peptide Design with Diversity-regularized Direct Preference Optimization
Oct 25, 2024Inverse folding models play an important role in structure-based design by predicting amino acid sequences that fold into desired reference structures. Models like ProteinMPNN, a message-passing encoder-decoder model, are trained to reliably produce new sequences from a reference structure. However, when applied to peptides, these models are prone to generating repetitive sequences that do not fold into the reference structure. To address this, we fine-tune ProteinMPNN to produce diverse and structurally consistent peptide sequences via Direct Preference Optimization (DPO). We derive two enhancements to DPO: online diversity regularization and domain-specific priors. Additionally, we develop a new understanding on improving diversity in decoder models. When conditioned on OpenFold generated structures, our fine-tuned models achieve state-of-the-art structural similarity scores, improving base ProteinMPNN by at least 8%. Compared to standard DPO, our regularized method achieves up to 20% higher sequence diversity with no loss in structural similarity score.