 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeJESTR: Joint Embedding Space Technique for Ranking Candidate Molecules for the Annotation of Untargeted Metabolomics Data
Nov 25, 2024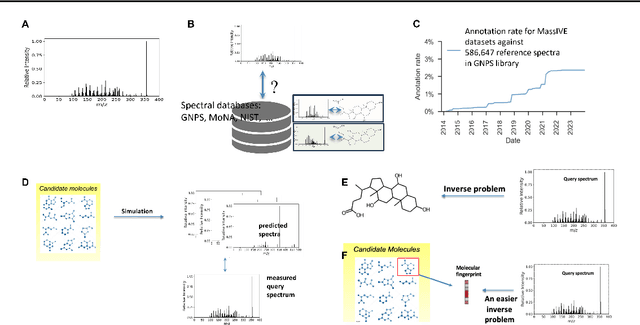
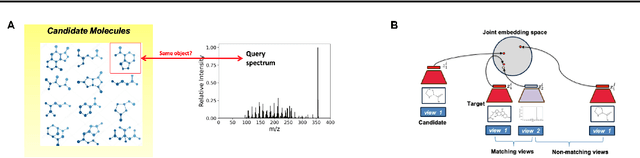

Motivation: A major challenge in metabolomics is annotation: assigning molecular structures to mass spectral fragmentation patterns. Despite recent advances in molecule-to-spectra and in spectra-to-molecular fingerprint prediction (FP), annotation rates remain low. Results: We introduce in this paper a novel paradigm (JESTR) for annotation. Unlike prior approaches that explicitly construct molecular fingerprints or spectra, JESTR leverages the insight that molecules and their corresponding spectra are views of the same data and effectively embeds their representations in a joint space. Candidate structures are ranked based on cosine similarity between the embeddings of query spectrum and each candidate. We evaluate JESTR against mol-to-spec and spec-to-FP annotation tools on three datasets. On average, for rank@[1-5], JESTR outperforms other tools by 23.6%-71.6%. We further demonstrate the strong value of regularization with candidate molecules during training, boosting rank@1 performance by 11.4% and enhancing the model's ability to discern between target and candidate molecules. Through JESTR, we offer a novel promising avenue towards accurate annotation, therefore unlocking valuable insights into the metabolome.
MassSpecGym: A benchmark for the discovery and identification of molecules
Oct 30, 2024The discovery and identification of molecules in biological and environmental samples is crucial for advancing biomedical and chemical sciences. Tandem mass spectrometry (MS/MS) is the leading technique for high-throughput elucidation of molecular structures. However, decoding a molecular structure from its mass spectrum is exceptionally challenging, even when performed by human experts. As a result, the vast majority of acquired MS/MS spectra remain uninterpreted, thereby limiting our understanding of the underlying (bio)chemical processes. Despite decades of progress in machine learning applications for predicting molecular structures from MS/MS spectra, the development of new methods is severely hindered by the lack of standard datasets and evaluation protocols. To address this problem, we propose MassSpecGym -- the first comprehensive benchmark for the discovery and identification of molecules from MS/MS data. Our benchmark comprises the largest publicly available collection of high-quality labeled MS/MS spectra and defines three MS/MS annotation challenges: \textit{de novo} molecular structure generation, molecule retrieval, and spectrum simulation. It includes new evaluation metrics and a generalization-demanding data split, therefore standardizing the MS/MS annotation tasks and rendering the problem accessible to the broad machine learning community. MassSpecGym is publicly available at \url{https://github.com/pluskal-lab/MassSpecGym}.
Contrastive Multiview Coding for Enzyme-Substrate Interaction Prediction
Nov 18, 2021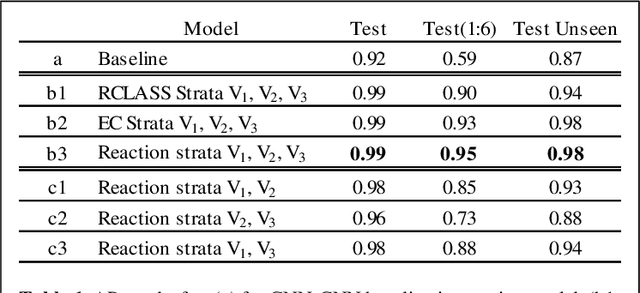
Characterizing Enzyme function is an important requirement for predicting Enzyme-Substrate interactions. In this paper, we present a novel approach of applying Contrastive Multiview Coding to this problem to improve the performance of prediction. We present a method to leverage auxiliary data from an Enzymatic database like KEGG to learn the mutual information present in multiple views of enzyme-substrate reactions. We show that congruency in the multiple views of the reaction data can be used to improve prediction performance.