 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI-Driven Expansion and Application of the Alexandria Database
Dec 09, 2025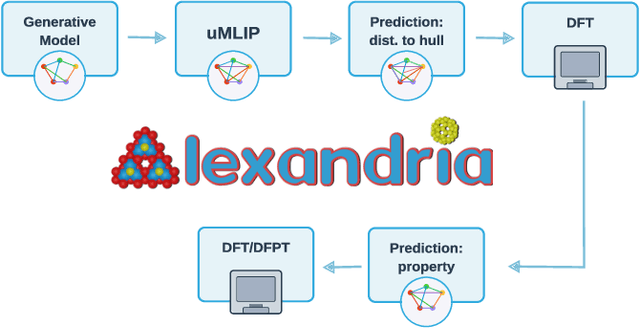
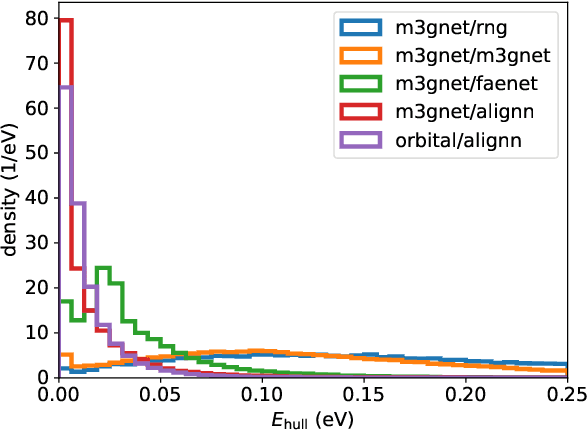
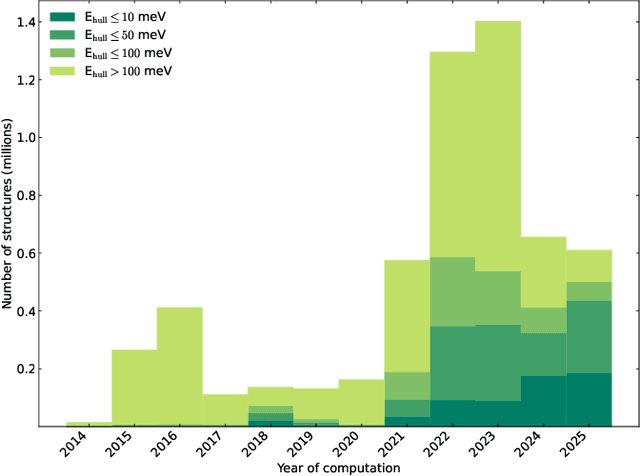
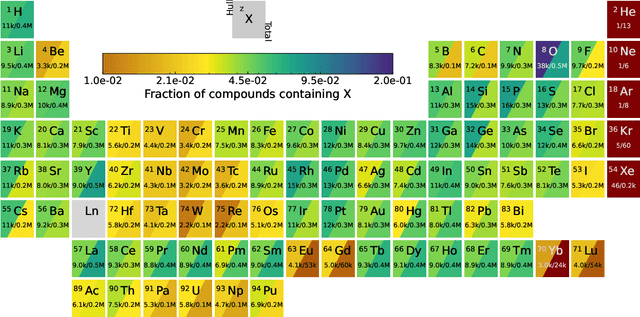
We present a novel multi-stage workflow for computational materials discovery that achieves a 99% success rate in identifying compounds within 100 meV/atom of thermodynamic stability, with a threefold improvement over previous approaches. By combining the Matra-Genoa generative model, Orb-v2 universal machine learning interatomic potential, and ALIGNN graph neural network for energy prediction, we generated 119 million candidate structures and added 1.3 million DFT-validated compounds to the ALEXANDRIA database, including 74 thousand new stable materials. The expanded ALEXANDRIA database now contains 5.8 million structures with 175 thousand compounds on the convex hull. Predicted structural disorder rates (37-43%) match experimental databases, unlike other recent AI-generated datasets. Analysis reveals fundamental patterns in space group distributions, coordination environments, and phase stability networks, including sub-linear scaling of convex hull connectivity. We release the complete dataset, including sAlex25 with 14 million out-of-equilibrium structures containing forces and stresses for training universal force fields. We demonstrate that fine-tuning a GRACE model on this data improves benchmark accuracy. All data, models, and workflows are freely available under Creative Commons licenses.
Supervised Pretraining for Material Property Prediction
Apr 27, 2025Accurate prediction of material properties facilitates the discovery of novel materials with tailored functionalities. Deep learning models have recently shown superior accuracy and flexibility in capturing structure-property relationships. However, these models often rely on supervised learning, which requires large, well-annotated datasets an expensive and time-consuming process. Self-supervised learning (SSL) offers a promising alternative by pretraining on large, unlabeled datasets to develop foundation models that can be fine-tuned for material property prediction. In this work, we propose supervised pretraining, where available class information serves as surrogate labels to guide learning, even when downstream tasks involve unrelated material properties. We evaluate this strategy on two state-of-the-art SSL models and introduce a novel framework for supervised pretraining. To further enhance representation learning, we propose a graph-based augmentation technique that injects noise to improve robustness without structurally deforming material graphs. The resulting foundation models are fine-tuned for six challenging material property predictions, achieving significant performance gains over baselines, ranging from 2% to 6.67% improvement in mean absolute error (MAE) and establishing a new benchmark in material property prediction. This study represents the first exploration of supervised pertaining with surrogate labels in material property prediction, advancing methodology and application in the field.
Enhancing material property prediction with ensemble deep graph convolutional networks
Jul 26, 2024



Machine learning (ML) models have emerged as powerful tools for accelerating materials discovery and design by enabling accurate predictions of properties from compositional and structural data. These capabilities are vital for developing advanced technologies across fields such as energy, electronics, and biomedicine, potentially reducing the time and resources needed for new material exploration and promoting rapid innovation cycles. Recent efforts have focused on employing advanced ML algorithms, including deep learning - based graph neural network, for property prediction. Additionally, ensemble models have proven to enhance the generalizability and robustness of ML and DL. However, the use of such ensemble strategies in deep graph networks for material property prediction remains underexplored. Our research provides an in-depth evaluation of ensemble strategies in deep learning - based graph neural network, specifically targeting material property prediction tasks. By testing the Crystal Graph Convolutional Neural Network (CGCNN) and its multitask version, MT-CGCNN, we demonstrated that ensemble techniques, especially prediction averaging, substantially improve precision beyond traditional metrics for key properties like formation energy per atom ($\Delta E^{f}$), band gap ($E_{g}$) and density ($\rho$) in 33,990 stable inorganic materials. These findings support the broader application of ensemble methods to enhance predictive accuracy in the field.