 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Geometric Deep Learning on Molecular Graphs
Paper and Code
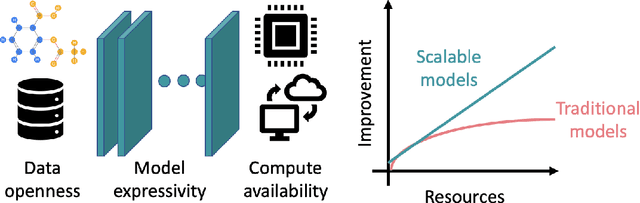

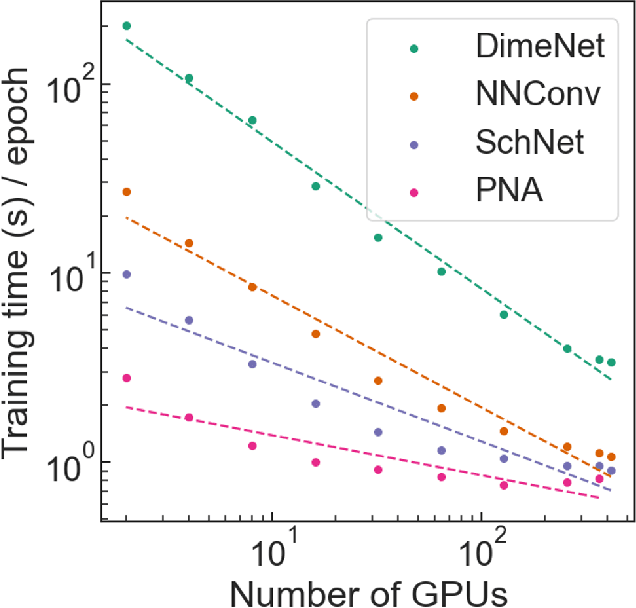
Deep learning in molecular and materials sciences is limited by the lack of integration between applied science, artificial intelligence, and high-performance computing. Bottlenecks with respect to the amount of training data, the size and complexity of model architectures, and the scale of the compute infrastructure are all key factors limiting the scaling of deep learning for molecules and materials. Here, we present $\textit{LitMatter}$, a lightweight framework for scaling molecular deep learning methods. We train four graph neural network architectures on over 400 GPUs and investigate the scaling behavior of these methods. Depending on the model architecture, training time speedups up to $60\times$ are seen. Empirical neural scaling relations quantify the model-dependent scaling and enable optimal compute resource allocation and the identification of scalable molecular geometric deep learning model implementations.