 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePan-cancer computational histopathology reveals tumor mutational burden status through weakly-supervised deep learning
Paper and Code
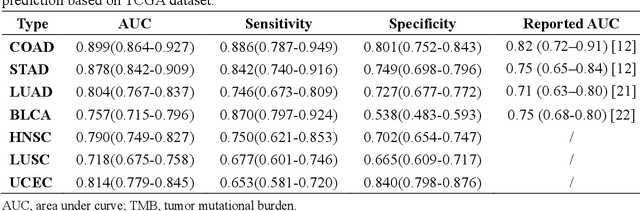
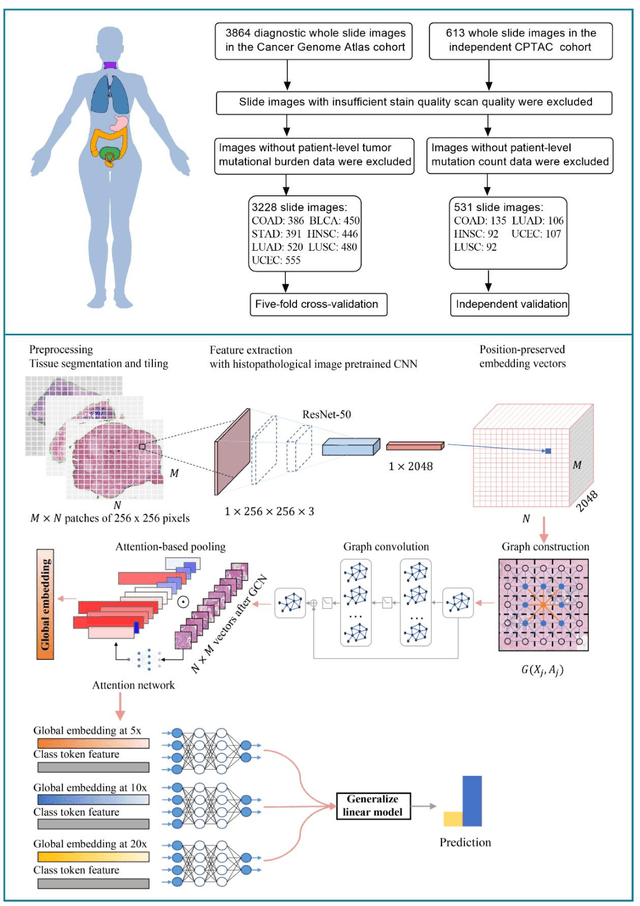
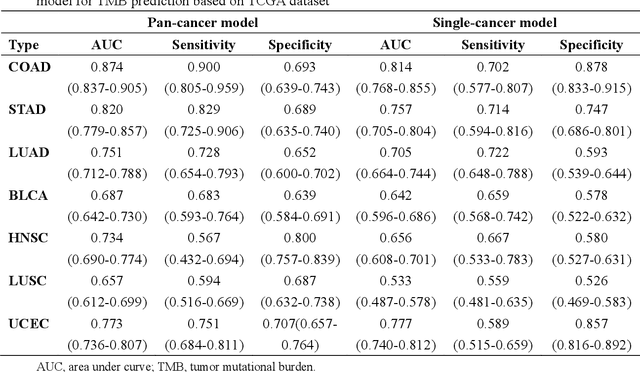
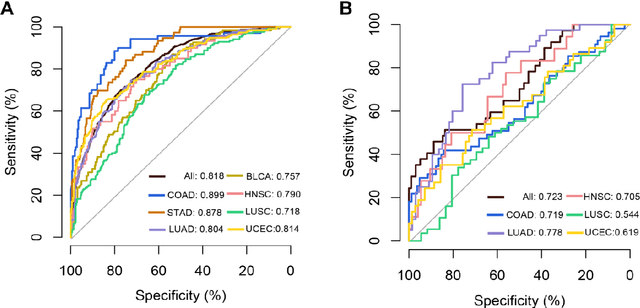
Tumor mutational burden (TMB) is a potential genomic biomarker that can help identify patients who will benefit from immunotherapy across a variety of cancers. We included whole slide images (WSIs) of 3228 diagnostic slides from the Cancer Genome Atlas and 531 WSIs from the Clinical Proteomic Tumor Analysis Consortium for the development and verification of a pan-cancer TMB prediction model (PC-TMB). We proposed a multiscale weakly-supervised deep learning framework for predicting TMB of seven types of tumors based only on routinely used hematoxylin-eosin (H&E)-stained WSIs. PC-TMB achieved a mean area under curve (AUC) of 0.818 (0.804-0.831) in the cross-validation cohort, which was superior to the best single-scale model. In comparison with the state-of-the-art TMB prediction model from previous publications, our multiscale model achieved better performance over previously reported models. In addition, the improvements of PC-TMB over the single-tumor models were also confirmed by the ablation tests on 10x magnification. The PC-TMB algorithm also exhibited good generalization on external validation cohort with AUC of 0.732 (0.683-0.761). PC-TMB possessed a comparable survival-risk stratification performance to the TMB measured by whole exome sequencing, but with low cost and being time-efficient for providing a prognostic biomarker of multiple solid tumors. Moreover, spatial heterogeneity of TMB within tumors was also identified through our PC-TMB, which might enable image-based screening for molecular biomarkers with spatial variation and potential exploring for genotype-spatial heterogeneity relationships.