 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecule Generation for Drug Design: a Graph Learning Perspective
Paper and Code
Feb 18, 2022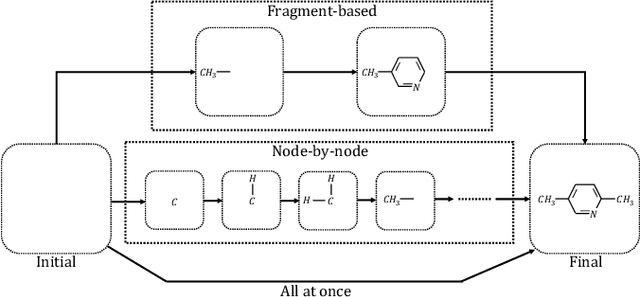
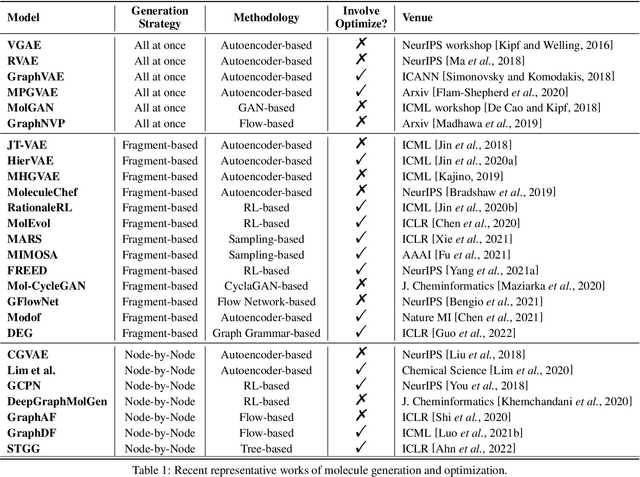
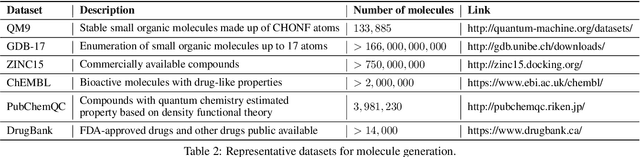
Machine learning has revolutionized many fields, and graph learning is recently receiving increasing attention. From the application perspective, one of the emerging and attractive areas is aiding the design and discovery of molecules, especially in drug industry. In this survey, we provide an overview of the state-of-the-art molecule (and mostly for de novo drug) design and discovery aiding methods whose methodology involves (deep) graph learning. Specifically, we propose to categorize these methods into three groups: i) all at once, ii) fragment-based and iii) node-by-node. We further present some representative public datasets and summarize commonly utilized evaluation metrics for generation and optimization, respectively. Finally, we discuss challenges and directions for future research, from the drug design perspective.