 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMetaRF: Differentiable Random Forest for Reaction Yield Prediction with a Few Trails
Paper and Code
Aug 22, 2022

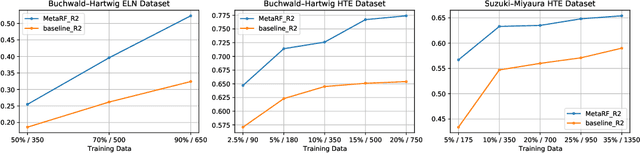
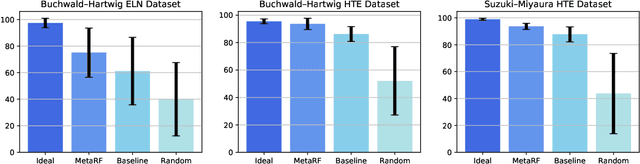
Artificial intelligence has deeply revolutionized the field of medicinal chemistry with many impressive applications, but the success of these applications requires a massive amount of training samples with high-quality annotations, which seriously limits the wide usage of data-driven methods. In this paper, we focus on the reaction yield prediction problem, which assists chemists in selecting high-yield reactions in a new chemical space only with a few experimental trials. To attack this challenge, we first put forth MetaRF, an attention-based differentiable random forest model specially designed for the few-shot yield prediction, where the attention weight of a random forest is automatically optimized by the meta-learning framework and can be quickly adapted to predict the performance of new reagents while given a few additional samples. To improve the few-shot learning performance, we further introduce a dimension-reduction based sampling method to determine valuable samples to be experimentally tested and then learned. Our methodology is evaluated on three different datasets and acquires satisfactory performance on few-shot prediction. In high-throughput experimentation (HTE) datasets, the average yield of our methodology's top 10 high-yield reactions is relatively close to the results of ideal yield selection.