 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeForce-matching Coarse-Graining without Forces
Paper and Code
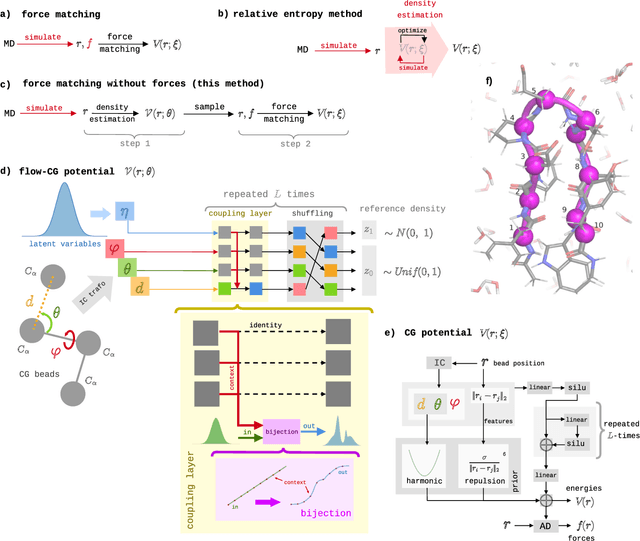
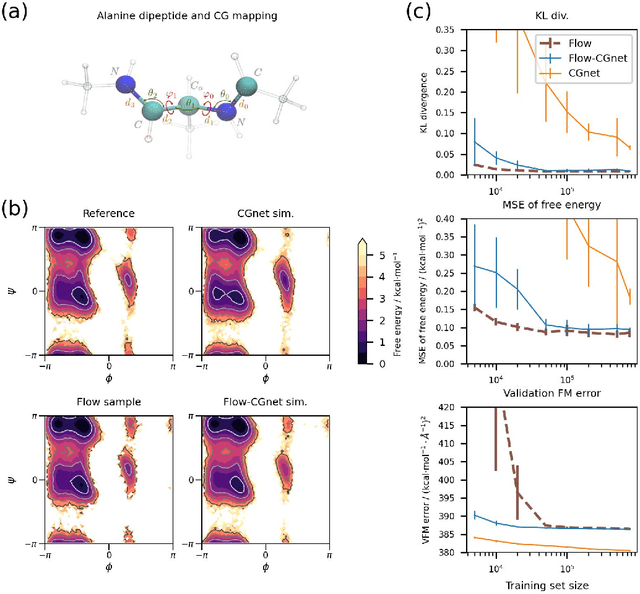
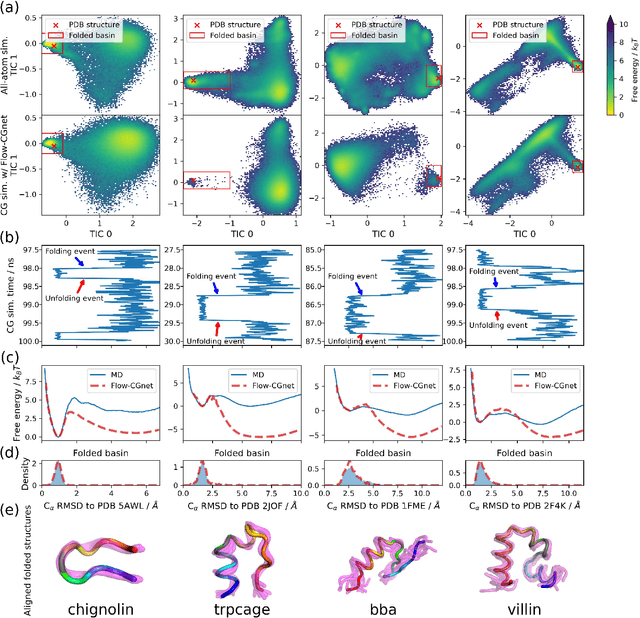
Coarse-grained (CG) molecular simulations have become a standard tool to study molecular processes on time-~and length-scales inaccessible to all-atom simulations. Learning CG force fields from all-atom data has mainly relied on force-matching and relative entropy minimization. Force-matching is straightforward to implement but requires the forces on the CG particles to be saved during all-atom simulation, and because these instantaneous forces depend on all degrees of freedom, they provide a very noisy signal that makes training the CG force field data inefficient. Relative entropy minimization does not require forces to be saved and is more data-efficient, but requires the CG model to be re-simulated during the iterative training procedure, which can make the training procedure extremely costly or lead to failure to converge. Here we present \emph{flow-matching}, a new training method for CG force fields that combines the advantages of force-matching and relative entropy minimization by leveraging normalizing flows, a generative deep learning method. Flow-matching first trains a normalizing flow to represent the CG probability density by using relative entropy minimization without suffering from the re-simulation problem because flows can directly sample from the equilibrium distribution they represent. Subsequently, the forces of the flow are used to train a CG force field by matching the coarse-grained forces directly, which is a much easier problem than traditional force-matching as it does not suffer from the noise problem. Besides not requiring forces, flow-matching also outperforms classical force-matching by an order of magnitude in terms of data efficiency and produces CG models that can capture the folding and unfolding of small proteins.