 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeContraNovo: A Contrastive Learning Approach to Enhance De Novo Peptide Sequencing
Paper and Code
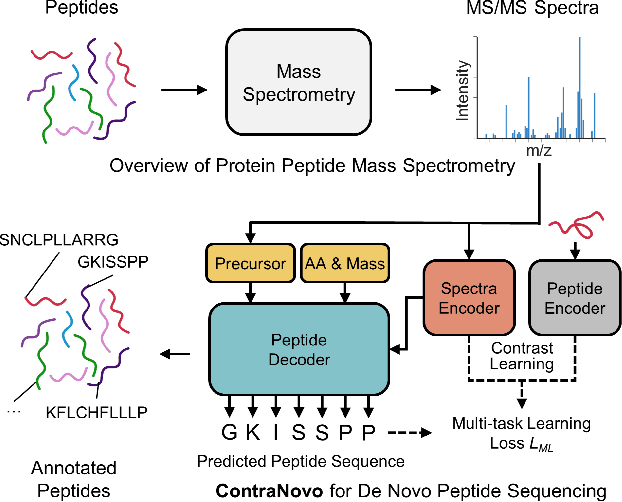
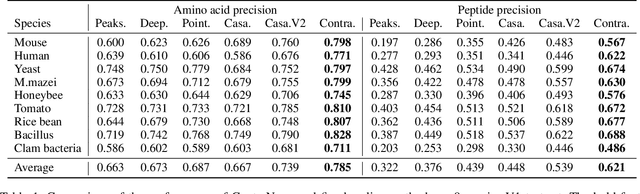
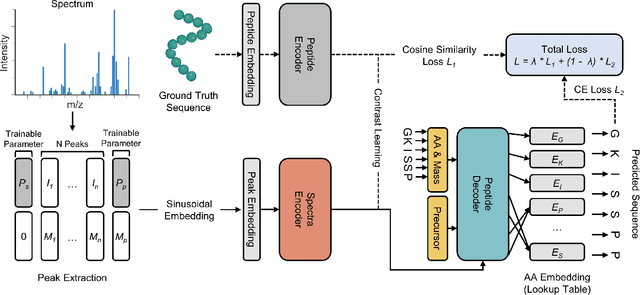

De novo peptide sequencing from mass spectrometry (MS) data is a critical task in proteomics research. Traditional de novo algorithms have encountered a bottleneck in accuracy due to the inherent complexity of proteomics data. While deep learning-based methods have shown progress, they reduce the problem to a translation task, potentially overlooking critical nuances between spectra and peptides. In our research, we present ContraNovo, a pioneering algorithm that leverages contrastive learning to extract the relationship between spectra and peptides and incorporates the mass information into peptide decoding, aiming to address these intricacies more efficiently. Through rigorous evaluations on two benchmark datasets, ContraNovo consistently outshines contemporary state-of-the-art solutions, underscoring its promising potential in enhancing de novo peptide sequencing. The source code is available at https://github.com/BEAM-Labs/ContraNovo.