 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoalescent-based species tree estimation: a stochastic Farris transform
Paper and Code
Jul 13, 2017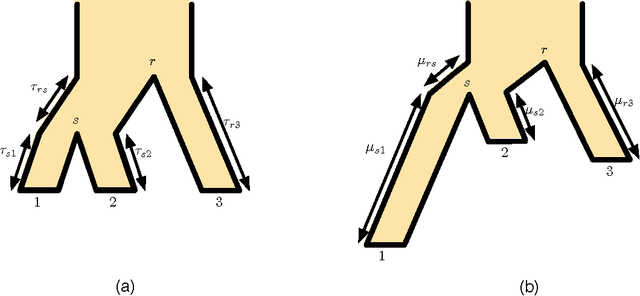

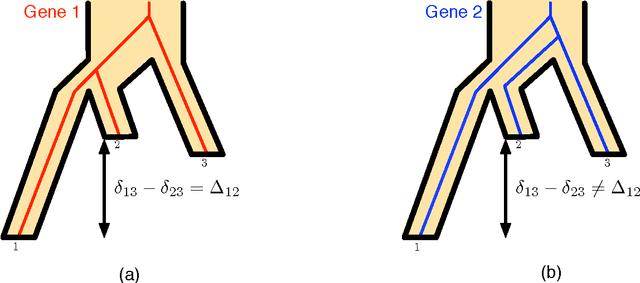
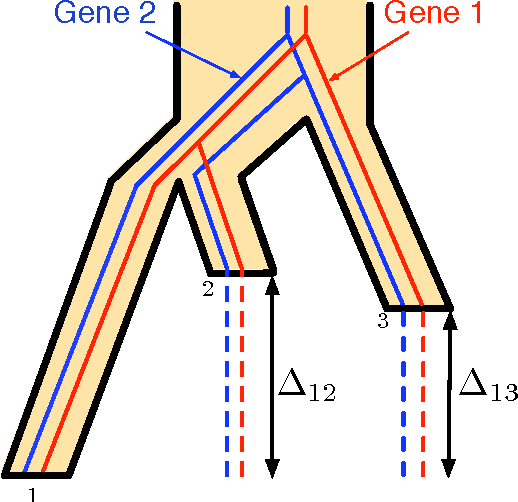
The reconstruction of a species phylogeny from genomic data faces two significant hurdles: 1) the trees describing the evolution of each individual gene--i.e., the gene trees--may differ from the species phylogeny and 2) the molecular sequences corresponding to each gene often provide limited information about the gene trees themselves. In this paper we consider an approach to species tree reconstruction that addresses both these hurdles. Specifically, we propose an algorithm for phylogeny reconstruction under the multispecies coalescent model with a standard model of site substitution. The multispecies coalescent is commonly used to model gene tree discordance due to incomplete lineage sorting, a well-studied population-genetic effect. In previous work, an information-theoretic trade-off was derived in this context between the number of loci, $m$, needed for an accurate reconstruction and the length of the locus sequences, $k$. It was shown that to reconstruct an internal branch of length $f$, one needs $m$ to be of the order of $1/[f^{2} \sqrt{k}]$. That previous result was obtained under the molecular clock assumption, i.e., under the assumption that mutation rates (as well as population sizes) are constant across the species phylogeny. Here we generalize this result beyond the restrictive molecular clock assumption, and obtain a new reconstruction algorithm that has the same data requirement (up to log factors). Our main contribution is a novel reduction to the molecular clock case under the multispecies coalescent. As a corollary, we also obtain a new identifiability result of independent interest: for any species tree with $n \geq 3$ species, the rooted species tree can be identified from the distribution of its unrooted weighted gene trees even in the absence of a molecular clock.