 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeomGCL: Geometric Graph Contrastive Learning for Molecular Property Prediction
Paper and Code
Sep 24, 2021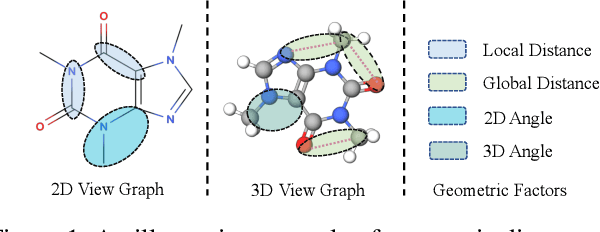
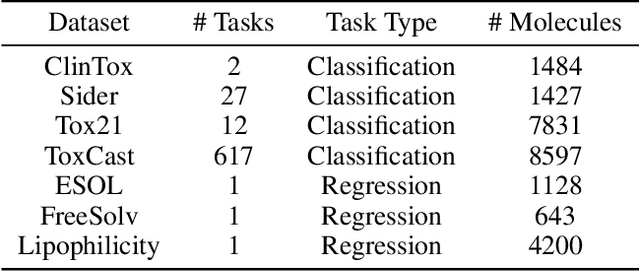
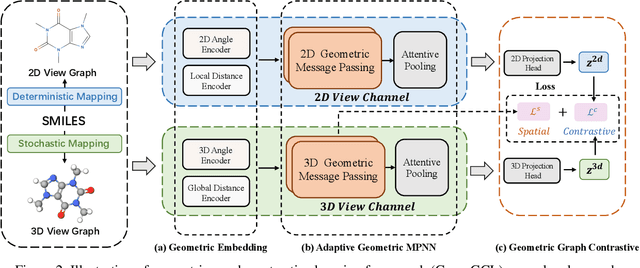
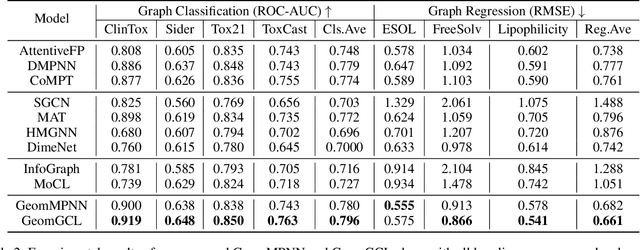
Recently many efforts have been devoted to applying graph neural networks (GNNs) to molecular property prediction which is a fundamental task for computational drug and material discovery. One of major obstacles to hinder the successful prediction of molecule property by GNNs is the scarcity of labeled data. Though graph contrastive learning (GCL) methods have achieved extraordinary performance with insufficient labeled data, most focused on designing data augmentation schemes for general graphs. However, the fundamental property of a molecule could be altered with the augmentation method (like random perturbation) on molecular graphs. Whereas, the critical geometric information of molecules remains rarely explored under the current GNN and GCL architectures. To this end, we propose a novel graph contrastive learning method utilizing the geometry of the molecule across 2D and 3D views, which is named GeomGCL. Specifically, we first devise a dual-view geometric message passing network (GeomMPNN) to adaptively leverage the rich information of both 2D and 3D graphs of a molecule. The incorporation of geometric properties at different levels can greatly facilitate the molecular representation learning. Then a novel geometric graph contrastive scheme is designed to make both geometric views collaboratively supervise each other to improve the generalization ability of GeomMPNN. We evaluate GeomGCL on various downstream property prediction tasks via a finetune process. Experimental results on seven real-life molecular datasets demonstrate the effectiveness of our proposed GeomGCL against state-of-the-art baselines.