 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Score-based Geometric Model for Molecular Dynamics Simulations
Paper and Code
Apr 19, 2022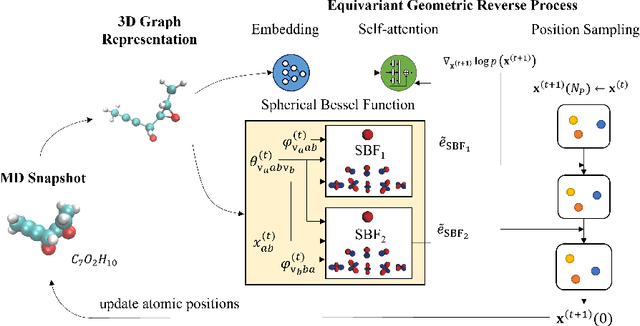
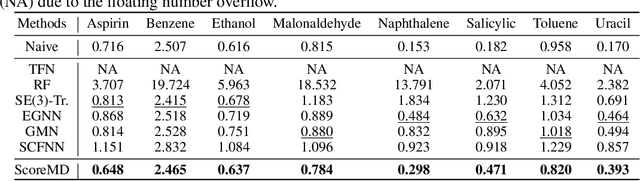
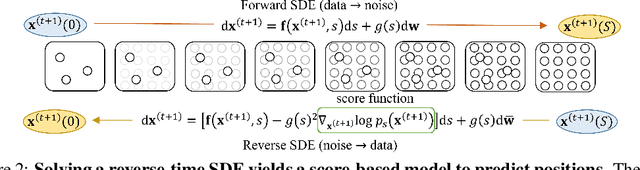

Molecular dynamics (MD) has long been the \emph{de facto} choice for modeling complex atomistic systems from first principles, and recently deep learning become a popular way to accelerate it. Notwithstanding, preceding approaches depend on intermediate variables such as the potential energy or force fields to update atomic positions, which requires additional computations to perform back-propagation. To waive this requirement, we propose a novel model called ScoreMD by directly estimating the gradient of the log density of molecular conformations. Moreover, we analyze that diffusion processes highly accord with the principle of enhanced sampling in MD simulations, and is therefore a perfect match to our sequential conformation generation task. That is, ScoreMD perturbs the molecular structure with a conditional noise depending on atomic accelerations and employs conformations at previous timeframes as the prior distribution for sampling. Another challenge of modeling such a conformation generation process is that the molecule is kinetic instead of static, which no prior studies strictly consider. To solve this challenge, we introduce a equivariant geometric Transformer as a score function in the diffusion process to calculate the corresponding gradient. It incorporates the directions and velocities of atomic motions via 3D spherical Fourier-Bessel representations. With multiple architectural improvements, we outperforms state-of-the-art baselines on MD17 and isomers of C7O2H10. This research provides new insights into the acceleration of new material and drug discovery.