 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular Dynamics Simulations on Cloud Computing and Machine Learning Platforms
Nov 11, 2021Scientific computing applications have benefited greatly from high performance computing infrastructure such as supercomputers. However, we are seeing a paradigm shift in the computational structure, design, and requirements of these applications. Increasingly, data-driven and machine learning approaches are being used to support, speed-up, and enhance scientific computing applications, especially molecular dynamics simulations. Concurrently, cloud computing platforms are increasingly appealing for scientific computing, providing "infinite" computing powers, easier programming and deployment models, and access to computing accelerators such as TPUs (Tensor Processing Units). This confluence of machine learning (ML) and cloud computing represents exciting opportunities for cloud and systems researchers. ML-assisted molecular dynamics simulations are a new class of workload, and exhibit unique computational patterns. These simulations present new challenges for low-cost and high-performance execution. We argue that transient cloud resources, such as low-cost preemptible cloud VMs, can be a viable platform for this new workload. Finally, we present some low-hanging fruits and long-term challenges in cloud resource management, and the integration of molecular dynamics simulations into ML platforms (such as TensorFlow).
Designing Machine Learning Surrogates using Outputs of Molecular Dynamics Simulations as Soft Labels
Oct 27, 2021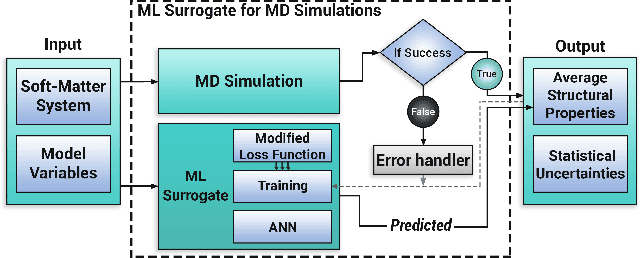
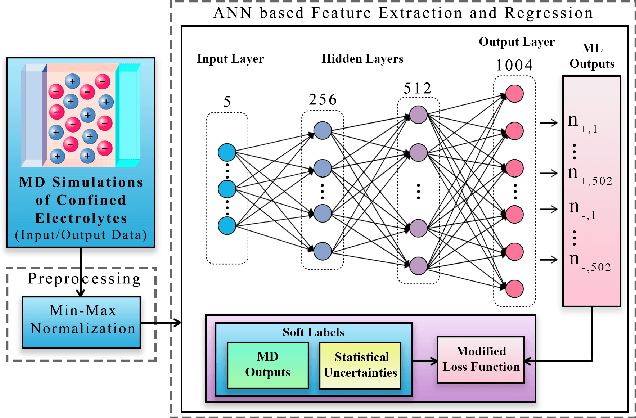
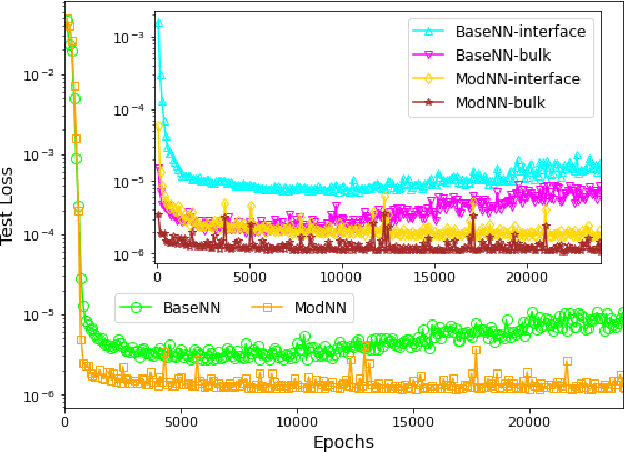
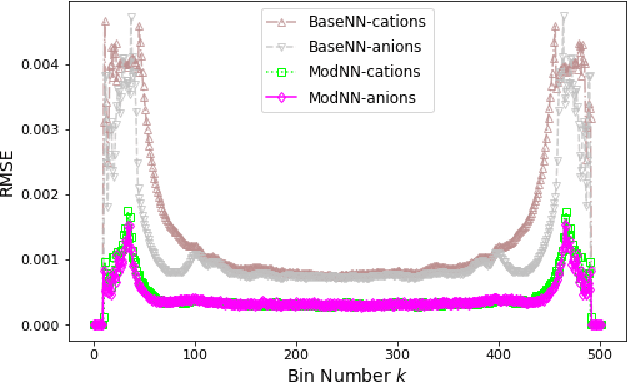
Molecular dynamics simulations are powerful tools to extract the microscopic mechanisms characterizing the properties of soft materials. We recently introduced machine learning surrogates for molecular dynamics simulations of soft materials and demonstrated that artificial neural network based regression models can successfully predict the relationships between the input material attributes and the simulation outputs. Here, we show that statistical uncertainties associated with the outputs of molecular dynamics simulations can be utilized to train artificial neural networks and design machine learning surrogates with higher accuracy and generalizability. We design soft labels for the simulation outputs by incorporating the uncertainties in the estimated average output quantities, and introduce a modified loss function that leverages these soft labels during training to significantly reduce the surrogate prediction error for input systems in the unseen test data. The approach is illustrated with the design of a surrogate for molecular dynamics simulations of confined electrolytes to predict the complex relationship between the input electrolyte attributes and the output ionic structure. The surrogate predictions for the ionic density profiles show excellent agreement with the ground truth results produced using molecular dynamics simulations. The high accuracy and small inference times associated with the surrogate predictions provide quick access to quantities derived using the number density profiles and facilitate rapid sensitivity analysis.
Deep Learning Based Integrators for Solving Newton's Equations with Large Timesteps
May 17, 2020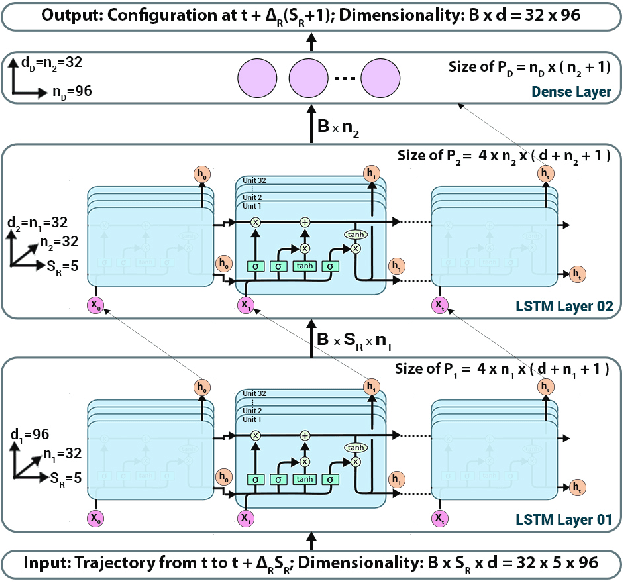
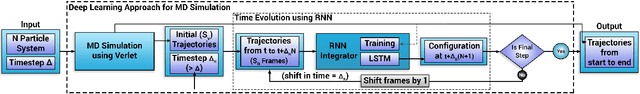
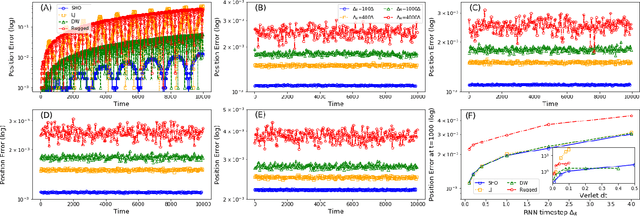
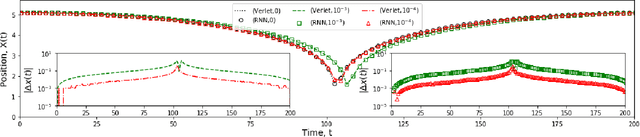
Classical molecular dynamics simulations are based on Newton's equations of motion and rely on numerical integrators to solve them. Using a small timestep to avoid discretization errors, Verlet integrators generate a trajectory of particle positions as solutions to Newton's equations. We introduce an integrator based on deep neural networks that is trained on trajectories generated using the Verlet integrator and learns to propagate the dynamics of particles with timestep up to 4000$\times$ larger compared to the Verlet timestep. We demonstrate significant net speedup of up to 32000 for 1 - 16 particle 3D systems and over a variety of force fields.