 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDesigning Machine Learning Surrogates using Outputs of Molecular Dynamics Simulations as Soft Labels
Paper and Code
Oct 27, 2021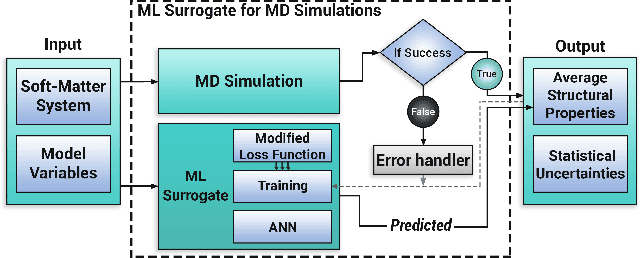
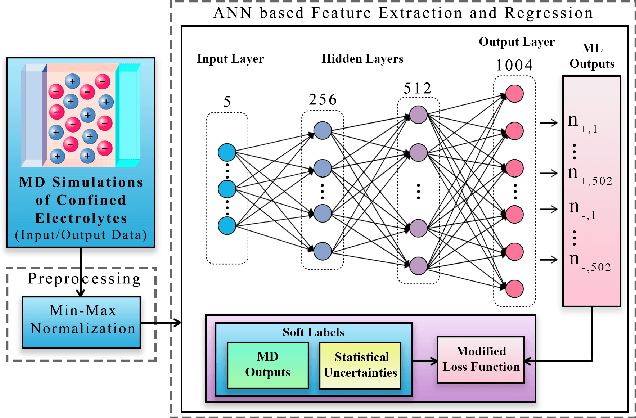
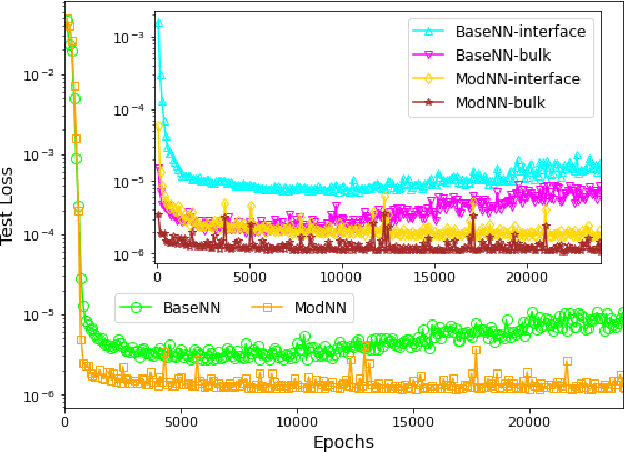
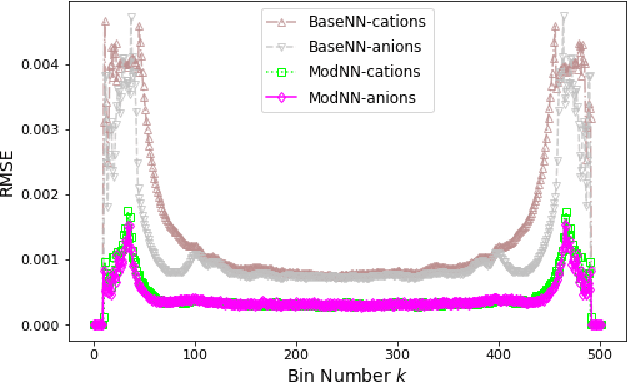
Molecular dynamics simulations are powerful tools to extract the microscopic mechanisms characterizing the properties of soft materials. We recently introduced machine learning surrogates for molecular dynamics simulations of soft materials and demonstrated that artificial neural network based regression models can successfully predict the relationships between the input material attributes and the simulation outputs. Here, we show that statistical uncertainties associated with the outputs of molecular dynamics simulations can be utilized to train artificial neural networks and design machine learning surrogates with higher accuracy and generalizability. We design soft labels for the simulation outputs by incorporating the uncertainties in the estimated average output quantities, and introduce a modified loss function that leverages these soft labels during training to significantly reduce the surrogate prediction error for input systems in the unseen test data. The approach is illustrated with the design of a surrogate for molecular dynamics simulations of confined electrolytes to predict the complex relationship between the input electrolyte attributes and the output ionic structure. The surrogate predictions for the ionic density profiles show excellent agreement with the ground truth results produced using molecular dynamics simulations. The high accuracy and small inference times associated with the surrogate predictions provide quick access to quantities derived using the number density profiles and facilitate rapid sensitivity analysis.