 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEffective and scalable clustering of SARS-CoV-2 sequences
Sep 10, 2021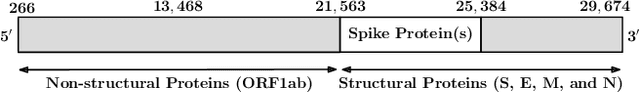
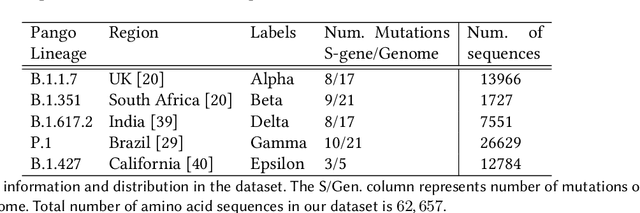
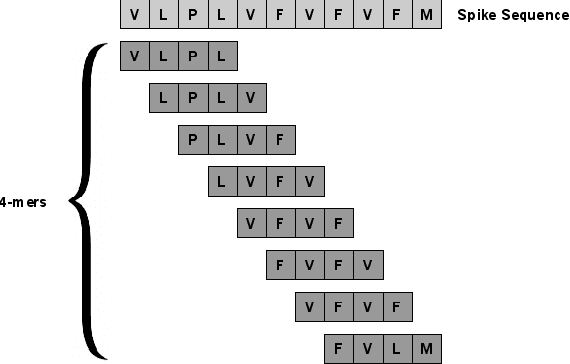
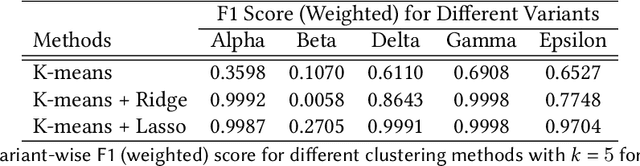
SARS-CoV-2, like any other virus, continues to mutate as it spreads, according to an evolutionary process. Unlike any other virus, the number of currently available sequences of SARS-CoV-2 in public databases such as GISAID is already several million. This amount of data has the potential to uncover the evolutionary dynamics of a virus like never before. However, a million is already several orders of magnitude beyond what can be processed by the traditional methods designed to reconstruct a virus's evolutionary history, such as those that build a phylogenetic tree. Hence, new and scalable methods will need to be devised in order to make use of the ever increasing number of viral sequences being collected. Since identifying variants is an important part of understanding the evolution of a virus, in this paper, we propose an approach based on clustering sequences to identify the current major SARS-CoV-2 variants. Using a $k$-mer based feature vector generation and efficient feature selection methods, our approach is effective in identifying variants, as well as being efficient and scalable to millions of sequences. Such a clustering method allows us to show the relative proportion of each variant over time, giving the rate of spread of each variant in different locations -- something which is important for vaccine development and distribution. We also compute the importance of each amino acid position of the spike protein in identifying a given variant in terms of information gain. Positions of high variant-specific importance tend to agree with those reported by the USA's Centers for Disease Control and Prevention (CDC), further demonstrating our approach.