 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQT-Net: Rethinking Evaluation of AI Models in Atomic Chemical Space
May 11, 2026Atomic properties such as partial charges or multipoles encode chemically meaningful information that can inform downstream molecular property prediction, but their evaluation as machine learning targets has been complicated by the absence of a principled out-of-distribution evaluation protocol at the atomic level. In this work, we propose a held-out evaluation protocol that clusters atomic environments by SOAP descriptors and computes metrics accounting only for cluster labels unseen during training. Following this procedure, we use 5$\times$5 cross-validation and Tukey's HSD to run a statistically rigorous comparison of E(3)-equivariant against non-equivariant, rotationally augmented models for predicting electron populations and multipoles of H, C, N, and O atoms. Building on our results, we introduce the Quantum Topological Neural Network (QT-Net), a rotationally augmented, non-equivariant graph neural network. We show that QT-Net can be used to infer properties of atoms in molecules from QM9 outside our training set, and that these inferred properties can yield improvement when used as input features for downstream molecular property prediction. To further validate the framework, molecular dipole moments computed from QT-Net's per-atom outputs recover the ground-truth values reported in QM9. We release all code and data, including a JAX implementation of QT-Net, to support the broader use of learned QTA properties as inductive biases for atomic-scale molecular machine learning.
deCIFer: Crystal Structure Prediction from Powder Diffraction Data using Autoregressive Language Models
Feb 04, 2025Novel materials drive progress across applications from energy storage to electronics. Automated characterization of material structures with machine learning methods offers a promising strategy for accelerating this key step in material design. In this work, we introduce an autoregressive language model that performs crystal structure prediction (CSP) from powder diffraction data. The presented model, deCIFer, generates crystal structures in the widely used Crystallographic Information File (CIF) format and can be conditioned on powder X-ray diffraction (PXRD) data. Unlike earlier works that primarily rely on high-level descriptors like composition, deCIFer performs CSP from diffraction data. We train deCIFer on nearly 2.3M unique crystal structures and validate on diverse sets of PXRD patterns for characterizing challenging inorganic crystal systems. Qualitative and quantitative assessments using the residual weighted profile and Wasserstein distance show that deCIFer produces structures that more accurately match the target diffraction data when conditioned, compared to the unconditioned case. Notably, deCIFer can achieve a 94% match rate on unseen data. deCIFer bridges experimental diffraction data with computational CSP, lending itself as a powerful tool for crystal structure characterization and accelerating materials discovery.
A Comprehensive Review of Emerging Approaches in Machine Learning for De Novo PROTAC Design
Jun 24, 2024Targeted protein degradation (TPD) is a rapidly growing field in modern drug discovery that aims to regulate the intracellular levels of proteins by harnessing the cell's innate degradation pathways to selectively target and degrade disease-related proteins. This strategy creates new opportunities for therapeutic intervention in cases where occupancy-based inhibitors have not been successful. Proteolysis-targeting chimeras (PROTACs) are at the heart of TPD strategies, which leverage the ubiquitin-proteasome system for the selective targeting and proteasomal degradation of pathogenic proteins. As the field evolves, it becomes increasingly apparent that the traditional methodologies for designing such complex molecules have limitations. This has led to the use of machine learning (ML) and generative modeling to improve and accelerate the development process. In this review, we explore the impact of ML on de novo PROTAC design $-$ an aspect of molecular design that has not been comprehensively reviewed despite its significance. We delve into the distinct characteristics of PROTAC linker design, underscoring the complexities required to create effective bifunctional molecules capable of TPD. We then examine how ML in the context of fragment-based drug design (FBDD), honed in the realm of small-molecule drug discovery, is paving the way for PROTAC linker design. Our review provides a critical evaluation of the limitations inherent in applying this method to the complex field of PROTAC development. Moreover, we review existing ML works applied to PROTAC design, highlighting pioneering efforts and, importantly, the limitations these studies face. By offering insights into the current state of PROTAC development and the integral role of ML in PROTAC design, we aim to provide valuable perspectives for researchers in their pursuit of better design strategies for this new modality.
De novo PROTAC design using graph-based deep generative models
Nov 04, 2022PROteolysis TArgeting Chimeras (PROTACs) are an emerging therapeutic modality for degrading a protein of interest (POI) by marking it for degradation by the proteasome. Recent developments in artificial intelligence (AI) suggest that deep generative models can assist with the de novo design of molecules with desired properties, and their application to PROTAC design remains largely unexplored. We show that a graph-based generative model can be used to propose novel PROTAC-like structures from empty graphs. Our model can be guided towards the generation of large molecules (30--140 heavy atoms) predicted to degrade a POI through policy-gradient reinforcement learning (RL). Rewards during RL are applied using a boosted tree surrogate model that predicts a molecule's degradation potential for each POI. Using this approach, we steer the generative model towards compounds with higher likelihoods of predicted degradation activity. Despite being trained on sparse public data, the generative model proposes molecules with substructures found in known degraders. After fine-tuning, predicted activity against a challenging POI increases from 50% to >80% with near-perfect chemical validity for sampled compounds, suggesting this is a promising approach for the optimization of large, PROTAC-like molecules for targeted protein degradation.
Amortized Tree Generation for Bottom-up Synthesis Planning and Synthesizable Molecular Design
Oct 12, 2021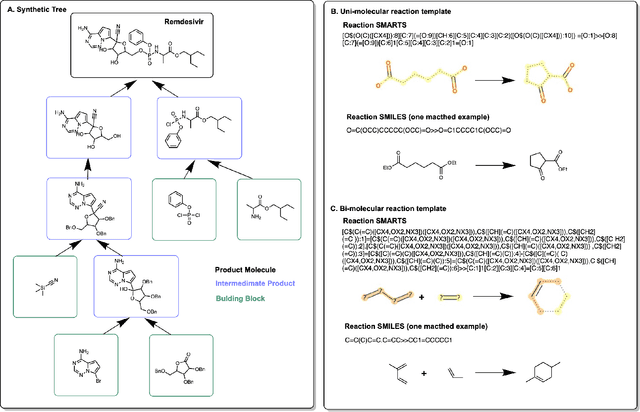
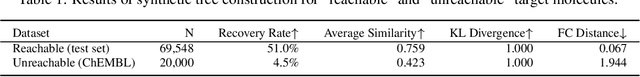

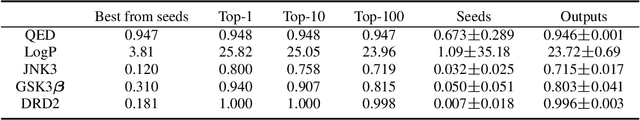
Molecular design and synthesis planning are two critical steps in the process of molecular discovery that we propose to formulate as a single shared task of conditional synthetic pathway generation. We report an amortized approach to generate synthetic pathways as a Markov decision process conditioned on a target molecular embedding. This approach allows us to conduct synthesis planning in a bottom-up manner and design synthesizable molecules by decoding from optimized conditional codes, demonstrating the potential to solve both problems of design and synthesis simultaneously. The approach leverages neural networks to probabilistically model the synthetic trees, one reaction step at a time, according to reactivity rules encoded in a discrete action space of reaction templates. We train these networks on hundreds of thousands of artificial pathways generated from a pool of purchasable compounds and a list of expert-curated templates. We validate our method with (a) the recovery of molecules using conditional generation, (b) the identification of synthesizable structural analogs, and (c) the optimization of molecular structures given oracle functions relevant to drug discovery.