 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDevelopment and Evaluation of Conformal Prediction Methods for QSAR
Apr 03, 2023


The quantitative structure-activity relationship (QSAR) regression model is a commonly used technique for predicting biological activities of compounds using their molecular descriptors. Predictions from QSAR models can help, for example, to optimize molecular structure; prioritize compounds for further experimental testing; and estimate their toxicity. In addition to the accurate estimation of the activity, it is highly desirable to obtain some estimate of the uncertainty associated with the prediction, e.g., calculate a prediction interval (PI) containing the true molecular activity with a pre-specified probability, say 70%, 90% or 95%. The challenge is that most machine learning (ML) algorithms that achieve superior predictive performance require some add-on methods for estimating uncertainty of their prediction. The development of these algorithms is an active area of research by statistical and ML communities but their implementation for QSAR modeling remains limited. Conformal prediction (CP) is a promising approach. It is agnostic to the prediction algorithm and can produce valid prediction intervals under some weak assumptions on the data distribution. We proposed computationally efficient CP algorithms tailored to the most advanced ML models, including Deep Neural Networks and Gradient Boosting Machines. The validity and efficiency of proposed conformal predictors are demonstrated on a diverse collection of QSAR datasets as well as simulation studies.
Light Gradient Boosting Machine as a Regression Method for Quantitative Structure-Activity Relationships
Apr 28, 2021
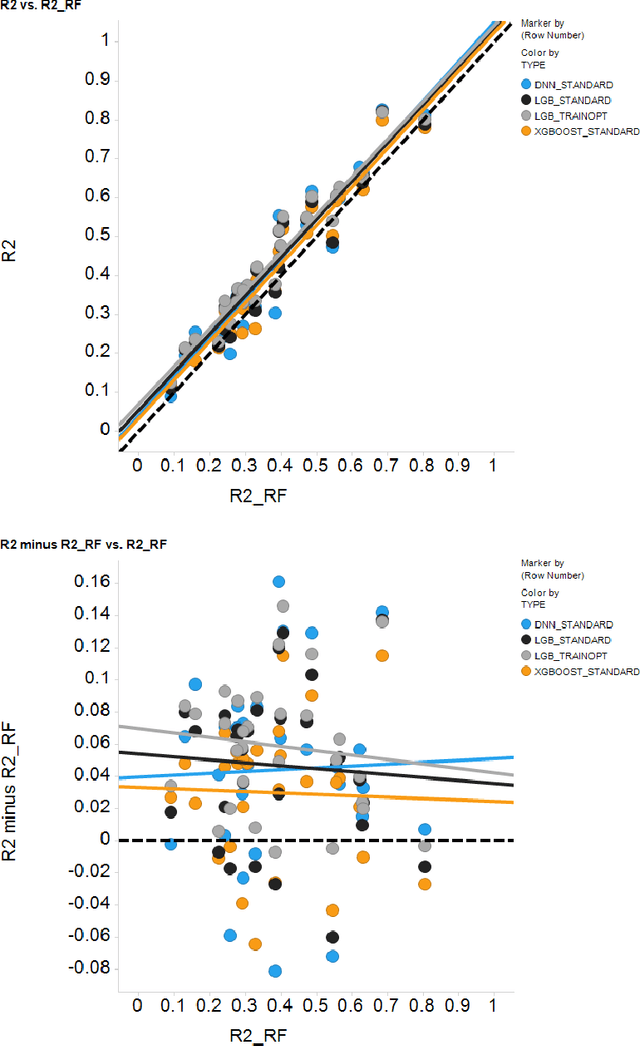
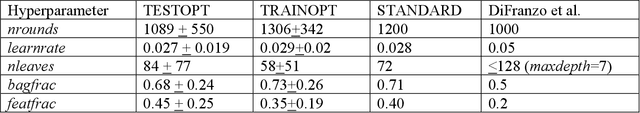
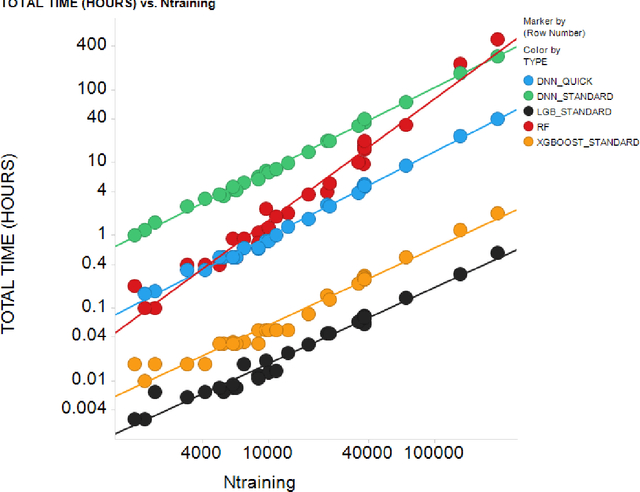
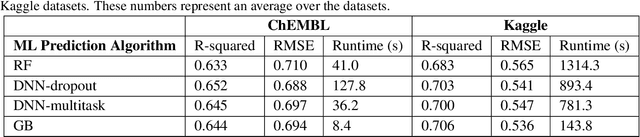
In the pharmaceutical industry, where it is common to generate many QSAR models with large numbers of molecules and descriptors, the best QSAR methods are those that can generate the most accurate predictions but that are also insensitive to hyperparameters and are computationally efficient. Here we compare Light Gradient Boosting Machine (LightGBM) to random forest, single-task deep neural nets, and Extreme Gradient Boosting (XGBoost) on 30 in-house data sets. While any boosting algorithm has many adjustable hyperparameters, we can define a set of standard hyperparameters at which LightGBM makes predictions about as accurate as single-task deep neural nets, but is a factor of 1000-fold faster than random forest and ~4-fold faster than XGBoost in terms of total computational time for the largest models. Another very useful feature of LightGBM is that it includes a native method for estimating prediction intervals.