 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBeyond Correlations: A Downstream Evaluation Framework for Query Performance Prediction
Jan 24, 2026The standard practice of query performance prediction (QPP) evaluation is to measure a set-level correlation between the estimated retrieval qualities and the true ones. However, neither this correlation-based evaluation measure quantifies QPP effectiveness at the level of individual queries, nor does this connect to a downstream application, meaning that QPP methods yielding high correlation values may not find a practical application in query-specific decisions in an IR pipeline. In this paper, we propose a downstream-focussed evaluation framework where a distribution of QPP estimates across a list of top-documents retrieved with several rankers is used as priors for IR fusion. While on the one hand, a distribution of these estimates closely matching that of the true retrieval qualities indicates the quality of the predictor, their usage as priors on the other hand indicates a predictor's ability to make informed choices in an IR pipeline. Our experiments firstly establish the importance of QPP estimates in weighted IR fusion, yielding substantial improvements of over 4.5% over unweighted CombSUM and RRF fusion strategies, and secondly, reveal new insights that the downstream effectiveness of QPP does not correlate well with the standard correlation-based QPP evaluation.
Breaking Flat: A Generalised Query Performance Prediction Evaluation Framework
Jan 24, 2026The traditional use-case of query performance prediction (QPP) is to identify which queries perform well and which perform poorly for a given ranking model. A more fine-grained and arguably more challenging extension of this task is to determine which ranking models are most effective for a given query. In this work, we generalize the QPP task and its evaluation into three settings: (i) SingleRanker MultiQuery (SRMQ-PP), corresponding to the standard use case; (ii) MultiRanker SingleQuery (MRSQ-PP), which evaluates a QPP model's ability to select the most effective ranker for a query; and (iii) MultiRanker MultiQuery (MRMQ-PP), which considers predictions jointly across all query ranker pairs. Our results show that (a) the relative effectiveness of QPP models varies substantially across tasks (SRMQ-PP vs. MRSQ-PP), and (b) predicting the best ranker for a query is considerably more difficult than predicting the relative difficulty of queries for a given ranker.
Unified Graph Networks (UGN): A Deep Neural Framework for Solving Graph Problems
Feb 11, 2025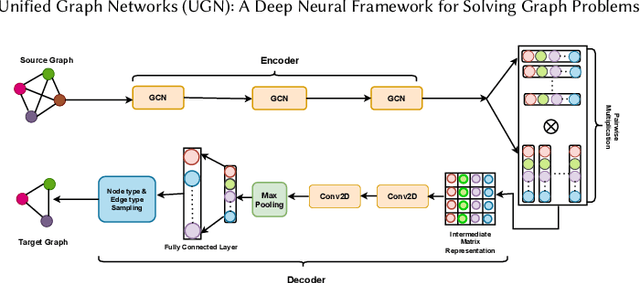
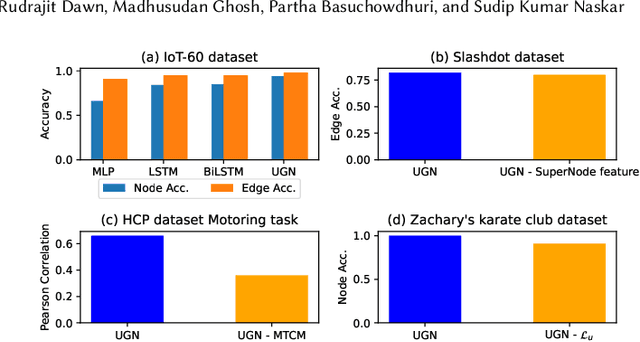
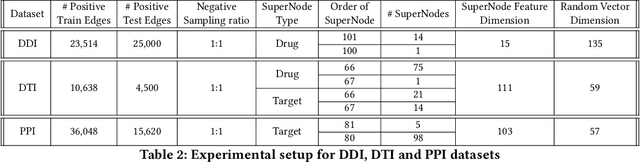
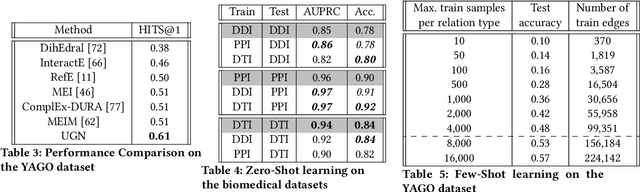
Deep neural networks have enabled researchers to create powerful generalized frameworks, such as transformers, that can be used to solve well-studied problems in various application domains, such as text and image. However, such generalized frameworks are not available for solving graph problems. Graph structures are ubiquitous in many applications around us and many graph problems have been widely studied over years. In recent times, there has been a surge in deep neural network based approaches to solve graph problems, with growing availability of graph structured datasets across diverse domains. Nevertheless, existing methods are mostly tailored to solve a specific task and lack the capability to create a generalized model leading to solutions for different downstream tasks. In this work, we propose a novel, resource-efficient framework named \emph{U}nified \emph{G}raph \emph{N}etwork (UGN) by leveraging the feature extraction capability of graph convolutional neural networks (GCN) and 2-dimensional convolutional neural networks (Conv2D). UGN unifies various graph learning tasks, such as link prediction, node classification, community detection, graph-to-graph translation, knowledge graph completion, and more, within a cohesive framework, while exercising minimal task-specific extensions (e.g., formation of supernodes for coarsening massive networks to increase scalability, use of \textit{mean target connectivity matrix} (MTCM) representation for achieving scalability in graph translation task, etc.) to enhance the generalization capability of graph learning and analysis. We test the novel UGN framework for six uncorrelated graph problems, using twelve different datasets. Experimental results show that UGN outperforms the state-of-the-art baselines by a significant margin on ten datasets, while producing comparable results on the remaining dataset.
AlpaPICO: Extraction of PICO Frames from Clinical Trial Documents Using LLMs
Sep 15, 2024



In recent years, there has been a surge in the publication of clinical trial reports, making it challenging to conduct systematic reviews. Automatically extracting Population, Intervention, Comparator, and Outcome (PICO) from clinical trial studies can alleviate the traditionally time-consuming process of manually scrutinizing systematic reviews. Existing approaches of PICO frame extraction involves supervised approach that relies on the existence of manually annotated data points in the form of BIO label tagging. Recent approaches, such as In-Context Learning (ICL), which has been shown to be effective for a number of downstream NLP tasks, require the use of labeled examples. In this work, we adopt ICL strategy by employing the pretrained knowledge of Large Language Models (LLMs), gathered during the pretraining phase of an LLM, to automatically extract the PICO-related terminologies from clinical trial documents in unsupervised set up to bypass the availability of large number of annotated data instances. Additionally, to showcase the highest effectiveness of LLM in oracle scenario where large number of annotated samples are available, we adopt the instruction tuning strategy by employing Low Rank Adaptation (LORA) to conduct the training of gigantic model in low resource environment for the PICO frame extraction task. Our empirical results show that our proposed ICL-based framework produces comparable results on all the version of EBM-NLP datasets and the proposed instruction tuned version of our framework produces state-of-the-art results on all the different EBM-NLP datasets. Our project is available at \url{https://github.com/shrimonmuke0202/AlpaPICO.git}.
CrysAtom: Distributed Representation of Atoms for Crystal Property Prediction
Sep 07, 2024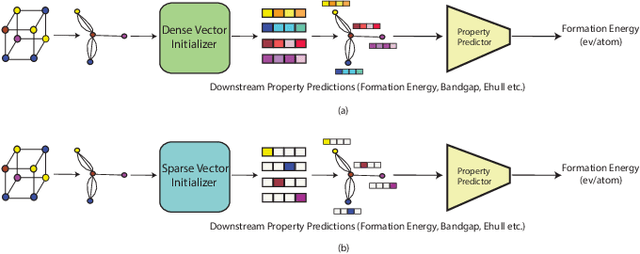
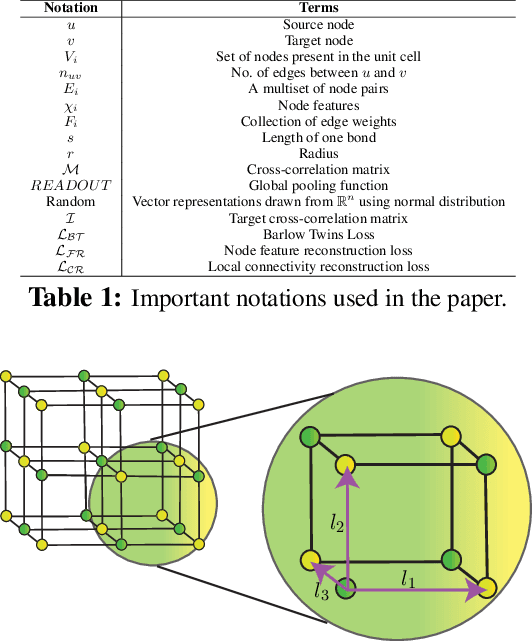
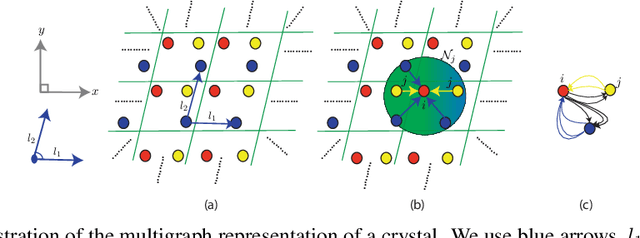
Application of artificial intelligence (AI) has been ubiquitous in the growth of research in the areas of basic sciences. Frequent use of machine learning (ML) and deep learning (DL) based methodologies by researchers has resulted in significant advancements in the last decade. These techniques led to notable performance enhancements in different tasks such as protein structure prediction, drug-target binding affinity prediction, and molecular property prediction. In material science literature, it is well-known that crystalline materials exhibit topological structures. Such topological structures may be represented as graphs and utilization of graph neural network (GNN) based approaches could help encoding them into an augmented representation space. Primarily, such frameworks adopt supervised learning techniques targeted towards downstream property prediction tasks on the basis of electronic properties (formation energy, bandgap, total energy, etc.) and crystalline structures. Generally, such type of frameworks rely highly on the handcrafted atom feature representations along with the structural representations. In this paper, we propose an unsupervised framework namely, CrysAtom, using untagged crystal data to generate dense vector representation of atoms, which can be utilized in existing GNN-based property predictor models to accurately predict important properties of crystals. Empirical results show that our dense representation embeds chemical properties of atoms and enhance the performance of the baseline property predictor models significantly.
G4-Attention: Deep Learning Model with Attention for predicting DNA G-Quadruplexes
Mar 05, 2024G-Quadruplexes are the four-stranded non-canonical nucleic acid secondary structures, formed by the stacking arrangement of the guanine tetramers. They are involved in a wide range of biological roles because of their exceptionally unique and distinct structural characteristics. After the completion of the human genome sequencing project, a lot of bioinformatic algorithms were introduced to predict the active G4s regions \textit{in vitro} based on the canonical G4 sequence elements, G-\textit{richness}, and G-\textit{skewness}, as well as the non-canonical sequence features. Recently, sequencing techniques like G4-seq and G4-ChIP-seq were developed to map the G4s \textit{in vitro}, and \textit{in vivo} respectively at a few hundred base resolution. Subsequently, several machine learning approaches were developed for predicting the G4 regions using the existing databases. However, their prediction models were simplistic, and the prediction accuracy was notably poor. In response, here, we propose a novel convolutional neural network with Bi-LSTM and attention layers, named G4-attention, to predict the G4 forming sequences with improved accuracy. G4-attention achieves high accuracy and attains state-of-the-art results in the G4 prediction task. Our model also predicts the G4 regions accurately in the highly class-imbalanced datasets. In addition, the developed model trained on the human genome dataset can be applied to any non-human genome DNA sequences to predict the G4 formation propensities.
Enhancing AI Research Paper Analysis: Methodology Component Extraction using Factored Transformer-based Sequence Modeling Approach
Nov 05, 2023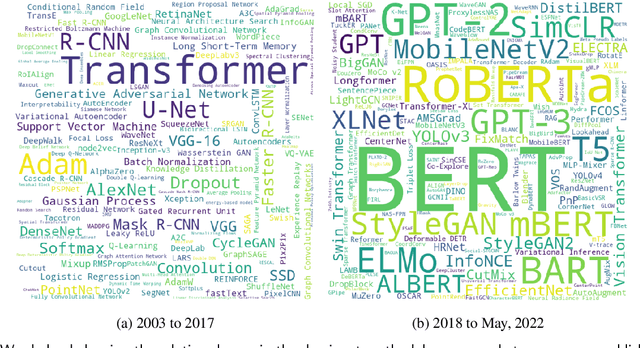
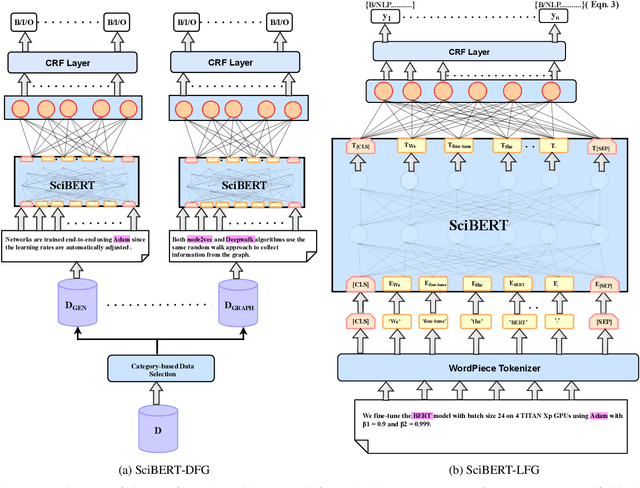

Research in scientific disciplines evolves, often rapidly, over time with the emergence of novel methodologies and their associated terminologies. While methodologies themselves being conceptual in nature and rather difficult to automatically extract and characterise, in this paper, we seek to develop supervised models for automatic extraction of the names of the various constituents of a methodology, e.g., `R-CNN', `ELMo' etc. The main research challenge for this task is effectively modeling the contexts around these methodology component names in a few-shot or even a zero-shot setting. The main contributions of this paper towards effectively identifying new evolving scientific methodology names are as follows: i) we propose a factored approach to sequence modeling, which leverages a broad-level category information of methodology domains, e.g., `NLP', `RL' etc.; ii) to demonstrate the feasibility of our proposed approach of identifying methodology component names under a practical setting of fast evolving AI literature, we conduct experiments following a simulated chronological setup (newer methodologies not seen during the training process); iii) our experiments demonstrate that the factored approach outperforms state-of-the-art baselines by margins of up to 9.257\% for the methodology extraction task with the few-shot setup.
Deep Graph Convolutional Network and LSTM based approach for predicting drug-target binding affinity
Jan 18, 2022Development of new drugs is an expensive and time-consuming process. Due to the world-wide SARS-CoV-2 outbreak, it is essential that new drugs for SARS-CoV-2 are developed as soon as possible. Drug repurposing techniques can reduce the time span needed to develop new drugs by probing the list of existing FDA-approved drugs and their properties to reuse them for combating the new disease. We propose a novel architecture DeepGLSTM, which is a Graph Convolutional network and LSTM based method that predicts binding affinity values between the FDA-approved drugs and the viral proteins of SARS-CoV-2. Our proposed model has been trained on Davis, KIBA (Kinase Inhibitor Bioactivity), DTC (Drug Target Commons), Metz, ToxCast and STITCH datasets. We use our novel architecture to predict a Combined Score (calculated using Davis and KIBA score) of 2,304 FDA-approved drugs against 5 viral proteins. On the basis of the Combined Score, we prepare a list of the top-18 drugs with the highest binding affinity for 5 viral proteins present in SARS-CoV-2. Subsequently, this list may be used for the creation of new useful drugs.