 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCrysAtom: Distributed Representation of Atoms for Crystal Property Prediction
Paper and Code
Sep 07, 2024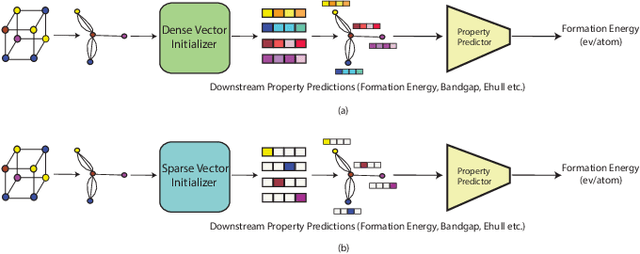
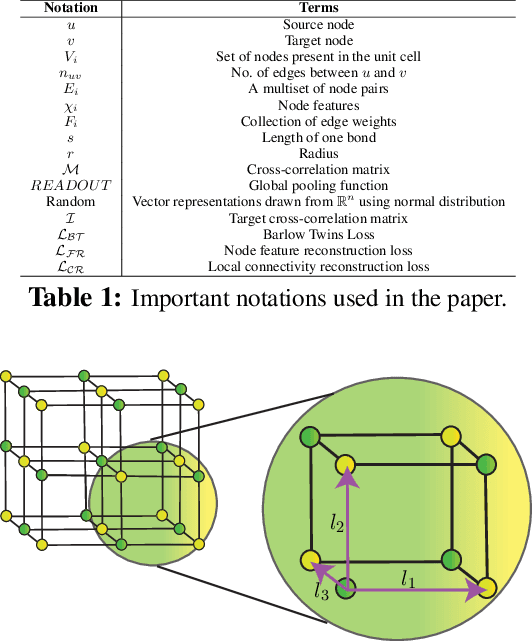
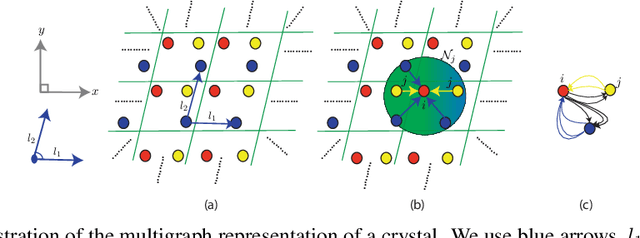
Application of artificial intelligence (AI) has been ubiquitous in the growth of research in the areas of basic sciences. Frequent use of machine learning (ML) and deep learning (DL) based methodologies by researchers has resulted in significant advancements in the last decade. These techniques led to notable performance enhancements in different tasks such as protein structure prediction, drug-target binding affinity prediction, and molecular property prediction. In material science literature, it is well-known that crystalline materials exhibit topological structures. Such topological structures may be represented as graphs and utilization of graph neural network (GNN) based approaches could help encoding them into an augmented representation space. Primarily, such frameworks adopt supervised learning techniques targeted towards downstream property prediction tasks on the basis of electronic properties (formation energy, bandgap, total energy, etc.) and crystalline structures. Generally, such type of frameworks rely highly on the handcrafted atom feature representations along with the structural representations. In this paper, we propose an unsupervised framework namely, CrysAtom, using untagged crystal data to generate dense vector representation of atoms, which can be utilized in existing GNN-based property predictor models to accurately predict important properties of crystals. Empirical results show that our dense representation embeds chemical properties of atoms and enhance the performance of the baseline property predictor models significantly.