 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMarkerMap: nonlinear marker selection for single-cell studies
Jul 28, 2022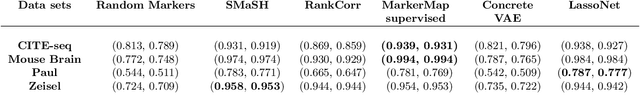
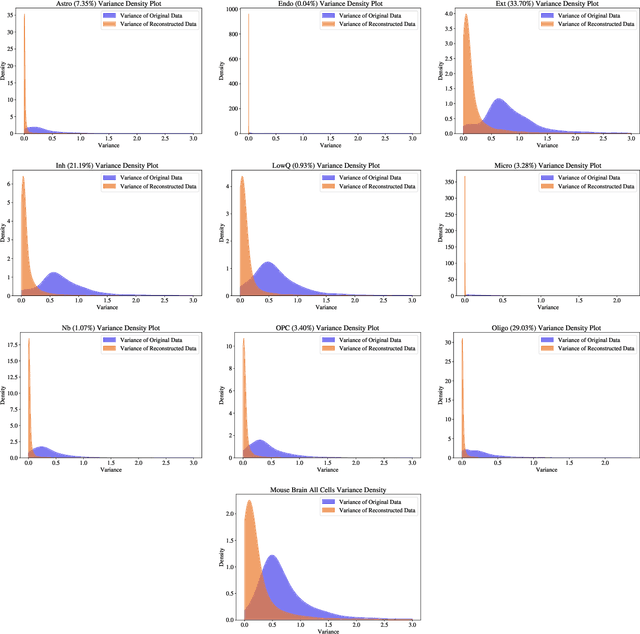
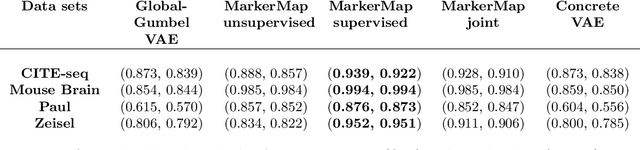
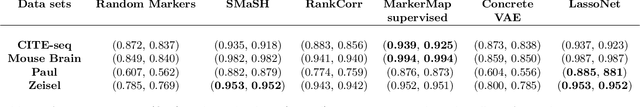
Single-cell RNA-seq data allow the quantification of cell type differences across a growing set of biological contexts. However, pinpointing a small subset of genomic features explaining this variability can be ill-defined and computationally intractable. Here we introduce MarkerMap, a generative model for selecting minimal gene sets which are maximally informative of cell type origin and enable whole transcriptome reconstruction. MarkerMap provides a scalable framework for both supervised marker selection, aimed at identifying specific cell type populations, and unsupervised marker selection, aimed at gene expression imputation and reconstruction. We benchmark MarkerMap's competitive performance against previously published approaches on real single cell gene expression data sets. MarkerMap is available as a pip installable package, as a community resource aimed at developing explainable machine learning techniques for enhancing interpretability in single-cell studies.