 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTechnical report: Improving the properties of molecules generated by LIMO
Jul 20, 2024This technical report investigates variants of the Latent Inceptionism on Molecules (LIMO) framework to improve the properties of generated molecules. We conduct ablative studies of molecular representation, decoder model, and surrogate model training scheme. The experiments suggest that an autogressive Transformer decoder with GroupSELFIES achieves the best average properties for the random generation task.
Target-Free Compound Activity Prediction via Few-Shot Learning
Nov 27, 2023Predicting the activities of compounds against protein-based or phenotypic assays using only a few known compounds and their activities is a common task in target-free drug discovery. Existing few-shot learning approaches are limited to predicting binary labels (active/inactive). However, in real-world drug discovery, degrees of compound activity are highly relevant. We study Few-Shot Compound Activity Prediction (FS-CAP) and design a novel neural architecture to meta-learn continuous compound activities across large bioactivity datasets. Our model aggregates encodings generated from the known compounds and their activities to capture assay information. We also introduce a separate encoder for the unknown compound. We show that FS-CAP surpasses traditional similarity-based techniques as well as other state of the art few-shot learning methods on a variety of target-free drug discovery settings and datasets.
LIMO: Latent Inceptionism for Targeted Molecule Generation
Jun 17, 2022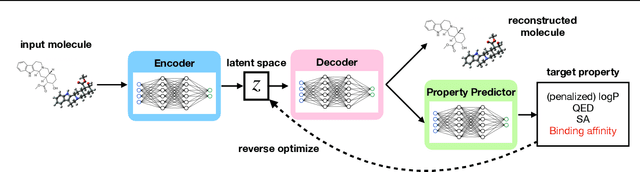
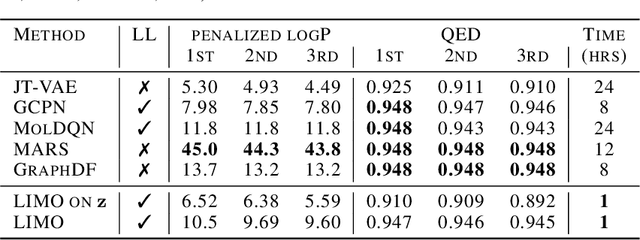
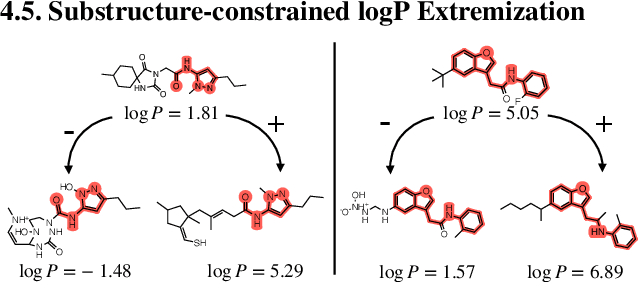

Generation of drug-like molecules with high binding affinity to target proteins remains a difficult and resource-intensive task in drug discovery. Existing approaches primarily employ reinforcement learning, Markov sampling, or deep generative models guided by Gaussian processes, which can be prohibitively slow when generating molecules with high binding affinity calculated by computationally-expensive physics-based methods. We present Latent Inceptionism on Molecules (LIMO), which significantly accelerates molecule generation with an inceptionism-like technique. LIMO employs a variational autoencoder-generated latent space and property prediction by two neural networks in sequence to enable faster gradient-based reverse-optimization of molecular properties. Comprehensive experiments show that LIMO performs competitively on benchmark tasks and markedly outperforms state-of-the-art techniques on the novel task of generating drug-like compounds with high binding affinity, reaching nanomolar range against two protein targets. We corroborate these docking-based results with more accurate molecular dynamics-based calculations of absolute binding free energy and show that one of our generated drug-like compounds has a predicted $K_D$ (a measure of binding affinity) of $6 \cdot 10^{-14}$ M against the human estrogen receptor, well beyond the affinities of typical early-stage drug candidates and most FDA-approved drugs to their respective targets. Code is available at https://github.com/Rose-STL-Lab/LIMO.