 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeApplication of Quaternions to Obtain Analytic Solutions to Systems of Polarization Components
May 21, 2025The usual way to describe mathematically a beam of coherent light passing through a system of waveplates is via the Jones vector and Jones matrix. This paper will show that a quaternion can be used to represent both the optical signal and the waveplate component it passes through, replacing the Jones vector and the Jones matrix. The quaternion description is easier to manipulate than the matrix-vector description; for example it can be inverted. As well as the Jones vector, the state of polarization (SOP) of an optical signal is often described as a three-dimensional vector on the Poincar\'e sphere, or as a polarization ellipse, and it will be shown how these three forms are closely related to the quaternion representation. Similarly, the action of a waveplate may be represented as a rotation about an axis on the Poincar\'e sphere, and that rotation is shown to have a logarithm-exponential relationship to the waveplate's quaternion. The paper presents rules to decide if two optical signals are aligned or orthogonal in phase or in polarization from their quaternions, and presents the quaternion operations to change the phase or change the SOP. Light passing through a system of waveplates is written as a product of quaternions, and it can be hard to simplify or manipulate that expression because quaternion multiplication does not commute. The paper brings together several mathematical tools that allow such a quaternion product to be rearranged, including the new idea of partial conjugation. Finally, a worked example is included of the quaternion mathematics applied to a waveplate problem that has not been solved before. It is shown that an endless optical phase shifter can be built using three rotatable waveplates, and equations for the angles of rotation are derived to produce the desired phase shift for given input and output SOPs.
Exploiting Ligand Additivity for Transferable Machine Learning of Multireference Character Across Known Transition Metal Complex Ligands
May 05, 2022
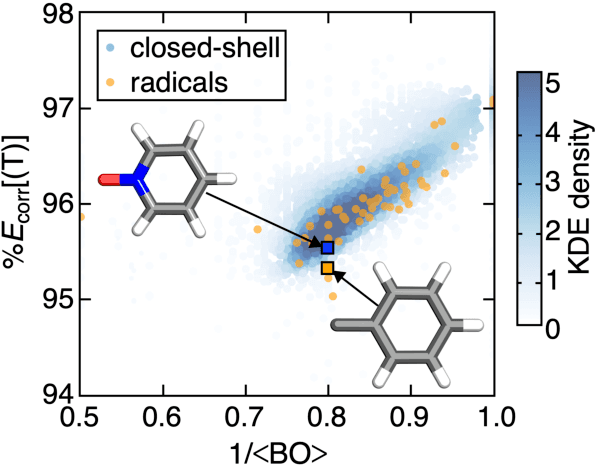

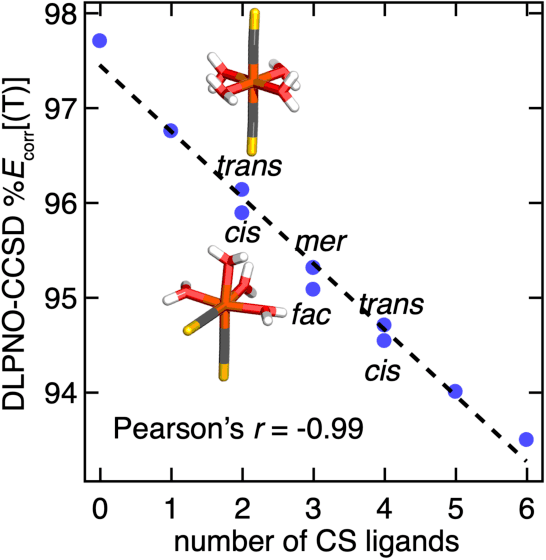
Accurate virtual high-throughput screening (VHTS) of transition metal complexes (TMCs) remains challenging due to the possibility of high multi-reference (MR) character that complicates property evaluation. We compute MR diagnostics for over 5,000 ligands present in previously synthesized transition metal complexes in the Cambridge Structural Database (CSD). To accomplish this task, we introduce an iterative approach for consistent ligand charge assignment for ligands in the CSD. Across this set, we observe that MR character correlates linearly with the inverse value of the averaged bond order over all bonds in the molecule. We then demonstrate that ligand additivity of MR character holds in TMCs, which suggests that the TMC MR character can be inferred from the sum of the MR character of the ligands. Encouraged by this observation, we leverage ligand additivity and develop a ligand-derived machine learning representation to train neural networks to predict the MR character of TMCs from properties of the constituent ligands. This approach yields models with excellent performance and superior transferability to unseen ligand chemistry and compositions.
Deciphering Cryptic Behavior in Bimetallic Transition Metal Complexes with Machine Learning
Jul 29, 2021



The rational tailoring of transition metal complexes is necessary to address outstanding challenges in energy utilization and storage. Heterobimetallic transition metal complexes that exhibit metal-metal bonding in stacked "double decker" ligand structures are an emerging, attractive platform for catalysis, but their properties are challenging to predict prior to laborious synthetic efforts. We demonstrate an alternative, data-driven approach to uncovering structure-property relationships for rational bimetallic complex design. We tailor graph-based representations of the metal-local environment for these heterobimetallic complexes for use in training of multiple linear regression and kernel ridge regression (KRR) models. Focusing on oxidation potentials, we obtain a set of 28 experimentally characterized complexes to develop a multiple linear regression model. On this training set, we achieve good accuracy (mean absolute error, MAE, of 0.25 V) and preserve transferability to unseen experimental data with a new ligand structure. We trained a KRR model on a subset of 330 structurally characterized heterobimetallics to predict the degree of metal-metal bonding. This KRR model predicts relative metal-metal bond lengths in the test set to within 5%, and analysis of key features reveals the fundamental atomic contributions (e.g., the valence electron configuration) that most strongly influence the behavior of complexes. Our work provides guidance for rational bimetallic design, suggesting that properties including the formal shortness ratio should be transferable from one period to another.
Machine learning to tame divergent density functional approximations: a new path to consensus materials design principles
Jun 24, 2021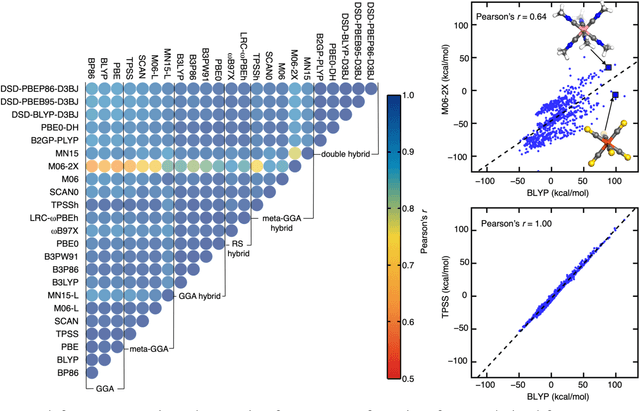


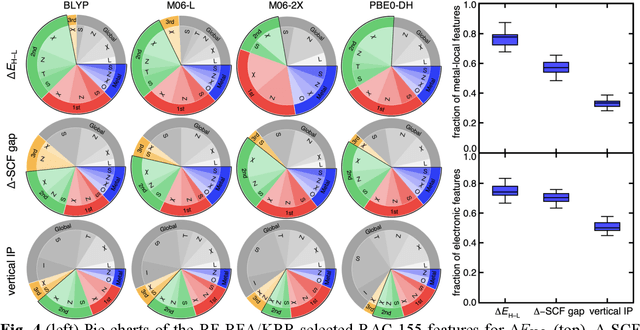
Computational virtual high-throughput screening (VHTS) with density functional theory (DFT) and machine-learning (ML)-acceleration is essential in rapid materials discovery. By necessity, efficient DFT-based workflows are carried out with a single density functional approximation (DFA). Nevertheless, properties evaluated with different DFAs can be expected to disagree for the cases with challenging electronic structure (e.g., open shell transition metal complexes, TMCs) for which rapid screening is most needed and accurate benchmarks are often unavailable. To quantify the effect of DFA bias, we introduce an approach to rapidly obtain property predictions from 23 representative DFAs spanning multiple families and "rungs" (e.g., semi-local to double hybrid) and basis sets on over 2,000 TMCs. Although computed properties (e.g., spin-state ordering and frontier orbital gap) naturally differ by DFA, high linear correlations persist across all DFAs. We train independent ML models for each DFA and observe convergent trends in feature importance; these features thus provide DFA-invariant, universal design rules. We devise a strategy to train ML models informed by all 23 DFAs and use them to predict properties (e.g., spin-splitting energy) of over 182k TMCs. By requiring consensus of the ANN-predicted DFA properties, we improve correspondence of these computational lead compounds with literature-mined, experimental compounds over the single-DFA approach typically employed. Both feature analysis and consensus-based ML provide efficient, alternative paths to overcome accuracy limitations of practical DFT.