 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDSVTLA: Deep Swin Vision Transformer-Based Transfer Learning Architecture for Multi-Type Cancer Histopathological Cancer Image Classification
Apr 10, 2026In this study, we proposed a deep Swin-Vision Transformer-based transfer learning architecture for robust multi-cancer histopathological image classification. The proposed framework integrates a hierarchical Swin Transformer with ResNet50-based convolution features extraction, enabling the model to capture both long-range contextual dependencies and fine-grained local morphological patterns within histopathological images. To validate the efficiency of the proposed architecture, an extensive experiment was executed on a comprehensive multi-cancer dataset including Breast Cancer, Oral Cancer, Lung and Colon Cancer, Kidney Cancer, and Acute Lymphocytic Leukemia (ALL), including both original and segmented images were analyzed to assess model robustness across heterogeneous clinical imaging conditions. Our approach is benchmarked alongside several state-of-the-art CNN and transfer models, including DenseNet121, DenseNet201, InceptionV3, ResNet50, EfficientNetB3, multiple ViT variants, and Swin Transformer models. However, all models were trained and validated using a unified pipeline, incorporating balanced data preprocessing, transfer learning, and fine-tuning strategies. The experimental results demonstrated that our proposed architecture consistently gained superior performance, reaching 100% test accuracy for lung-colon cancer, segmented leukemia datasets, and up to 99.23% accuracy for breast cancer classification. The model also achieved near-perfect precision, f1 score, and recall, indicating highly stable scores across divers cancer types. Overall, the proposed model establishes a highly accurate, interpretable, and also robust multi-cancer classification system, demonstrating strong benchmark for future research and provides a unified comparative assessment useful for designing reliable AI-assisted histopathological diagnosis and clinical decision-making.
Colorectal Cancer Histopathological Grading using Multi-Scale Federated Learning
Nov 05, 2025Colorectal cancer (CRC) grading is a critical prognostic factor but remains hampered by inter-observer variability and the privacy constraints of multi-institutional data sharing. While deep learning offers a path to automation, centralized training models conflict with data governance regulations and neglect the diagnostic importance of multi-scale analysis. In this work, we propose a scalable, privacy-preserving federated learning (FL) framework for CRC histopathological grading that integrates multi-scale feature learning within a distributed training paradigm. Our approach employs a dual-stream ResNetRS50 backbone to concurrently capture fine-grained nuclear detail and broader tissue-level context. This architecture is integrated into a robust FL system stabilized using FedProx to mitigate client drift across heterogeneous data distributions from multiple hospitals. Extensive evaluation on the CRC-HGD dataset demonstrates that our framework achieves an overall accuracy of 83.5%, outperforming a comparable centralized model (81.6%). Crucially, the system excels in identifying the most aggressive Grade III tumors with a high recall of 87.5%, a key clinical priority to prevent dangerous false negatives. Performance further improves with higher magnification, reaching 88.0% accuracy at 40x. These results validate that our federated multi-scale approach not only preserves patient privacy but also enhances model performance and generalization. The proposed modular pipeline, with built-in preprocessing, checkpointing, and error handling, establishes a foundational step toward deployable, privacy-aware clinical AI for digital pathology.
Weighted Scaling Approach for Metabolomics Data Analysis
Aug 01, 2022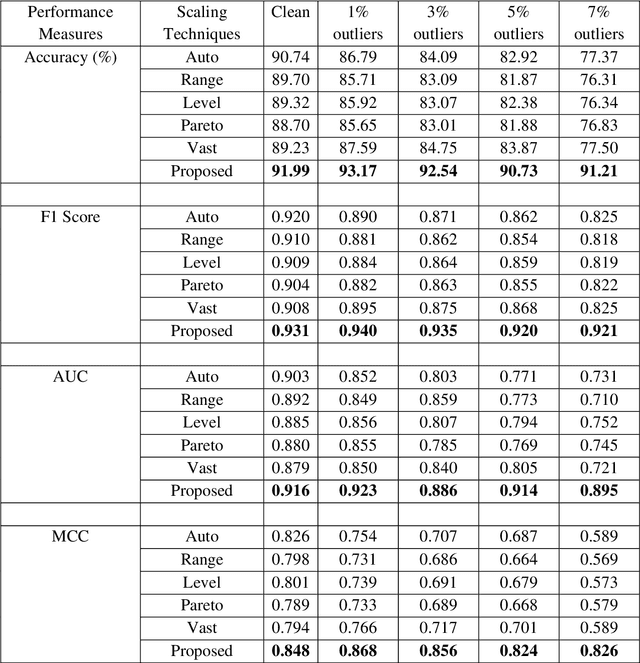


Systematic variation is a common issue in metabolomics data analysis. Therefore, different scaling and normalization techniques are used to preprocess the data for metabolomics data analysis. Although several scaling methods are available in the literature, however, choice of scaling, transformation and/or normalization technique influence the further statistical analysis. It is challenging to choose the appropriate scaling technique for downstream analysis to get accurate results or to make a proper decision. Moreover, the existing scaling techniques are sensitive to outliers or extreme values. To fill the gap, our objective is to introduce a robust scaling approach that is not influenced by outliers as well as provides more accurate results for downstream analysis. Here, we introduced a new weighted scaling approach that is robust against outliers however, where no additional outlier detection/treatment step is needed in data preprocessing and also compared it with the conventional scaling and normalization techniques through artificial and real metabolomics datasets. We evaluated the performance of the proposed method in comparison to the other existing conventional scaling techniques using metabolomics data analysis in both the absence and presence of different percentages of outliers. Results show that in most cases, the proposed scaling technique performs better than the traditional scaling methods in both the absence and presence of outliers. The proposed method improves the further downstream metabolomics analysis. The R function of the proposed robust scaling method is available at https://github.com/nishithkumarpaul/robustScaling/blob/main/wscaling.R
A robust kernel machine regression towards biomarker selection in multi-omics datasets of osteoporosis for drug discovery
Jan 13, 2022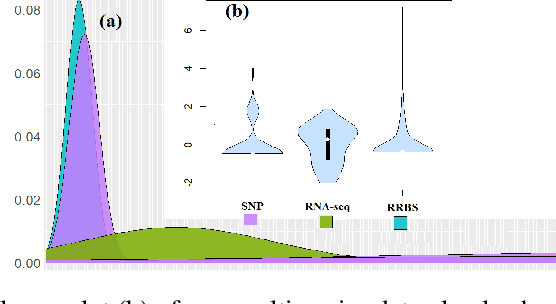
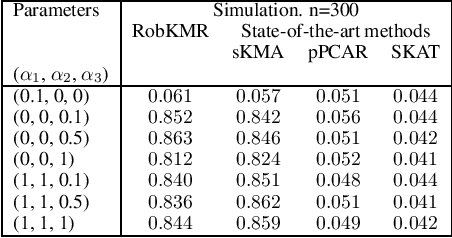
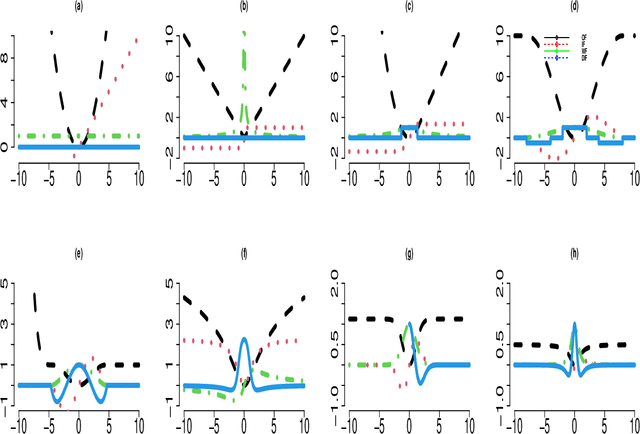
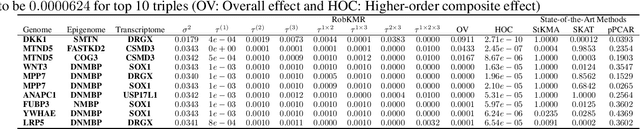
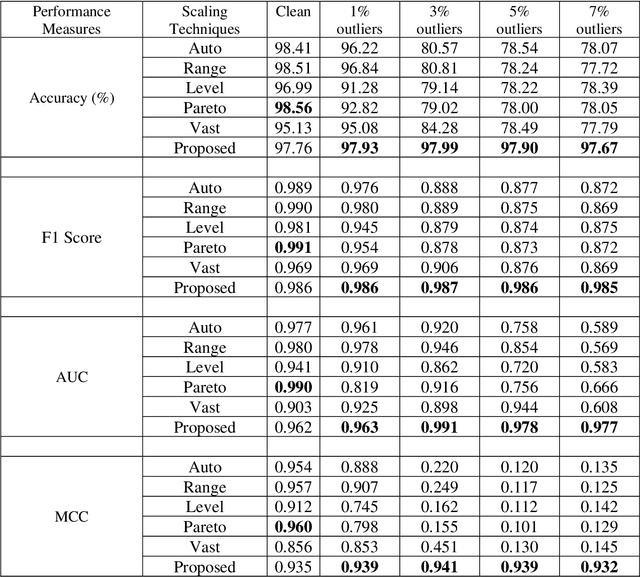
Many statistical machine approaches could ultimately highlight novel features of the etiology of complex diseases by analyzing multi-omics data. However, they are sensitive to some deviations in distribution when the observed samples are potentially contaminated with adversarial corrupted outliers (e.g., a fictional data distribution). Likewise, statistical advances lag in supporting comprehensive data-driven analyses of complex multi-omics data integration. We propose a novel non-linear M-estimator-based approach, "robust kernel machine regression (RobKMR)," to improve the robustness of statistical machine regression and the diversity of fictional data to examine the higher-order composite effect of multi-omics datasets. We address a robust kernel-centered Gram matrix to estimate the model parameters accurately. We also propose a robust score test to assess the marginal and joint Hadamard product of features from multi-omics data. We apply our proposed approach to a multi-omics dataset of osteoporosis (OP) from Caucasian females. Experiments demonstrate that the proposed approach effectively identifies the inter-related risk factors of OP. With solid evidence (p-value = 0.00001), biological validations, network-based analysis, causal inference, and drug repurposing, the selected three triplets ((DKK1, SMTN, DRGX), (MTND5, FASTKD2, CSMD3), (MTND5, COG3, CSMD3)) are significant biomarkers and directly relate to BMD. Overall, the top three selected genes (DKK1, MTND5, FASTKD2) and one gene (SIDT1 at p-value= 0.001) significantly bond with four drugs- Tacrolimus, Ibandronate, Alendronate, and Bazedoxifene out of 30 candidates for drug repurposing in OP. Further, the proposed approach can be applied to any disease model where multi-omics datasets are available.
A generalized kernel machine approach to identify higher-order composite effects in multi-view datasets
Apr 29, 2020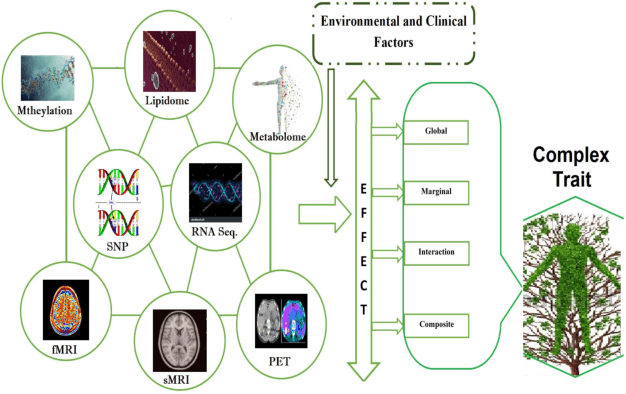

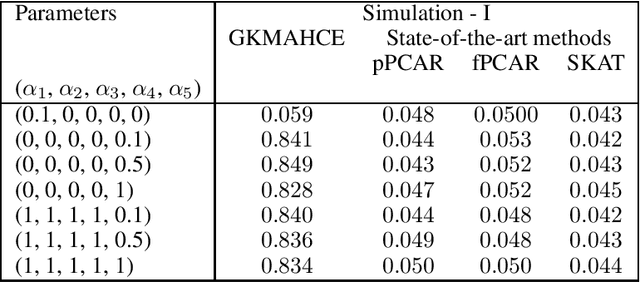
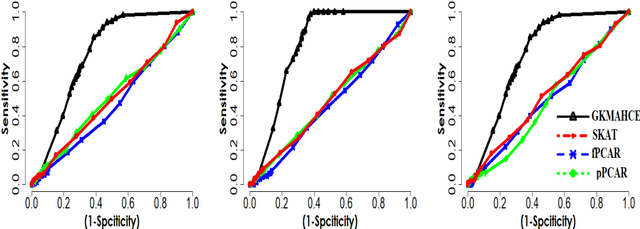
In recent years, a comprehensive study of multi-view datasets (e.g., multi-omics and imaging scans) has been a focus and forefront in biomedical research. State-of-the-art biomedical technologies are enabling us to collect multi-view biomedical datasets for the study of complex diseases. While all the views of data tend to explore complementary information of a disease, multi-view data analysis with complex interactions is challenging for a deeper and holistic understanding of biological systems. In this paper, we propose a novel generalized kernel machine approach to identify higher-order composite effects in multi-view biomedical datasets. This generalized semi-parametric (a mixed-effect linear model) approach includes the marginal and joint Hadamard product of features from different views of data. The proposed kernel machine approach considers multi-view data as predictor variables to allow more thorough and comprehensive modeling of a complex trait. The proposed method can be applied to the study of any disease model, where multi-view datasets are available. We applied our approach to both synthesized datasets and real multi-view datasets from adolescence brain development and osteoporosis study, including an imaging scan dataset and five omics datasets. Our experiments demonstrate that the proposed method can effectively identify higher-order composite effects and suggest that corresponding features (genes, region of interests, and chemical taxonomies) function in a concerted effort. We show that the proposed method is more generalizable than existing ones.
Identifying Outliers using Influence Function of Multiple Kernel Canonical Correlation Analysis
Jun 01, 2016
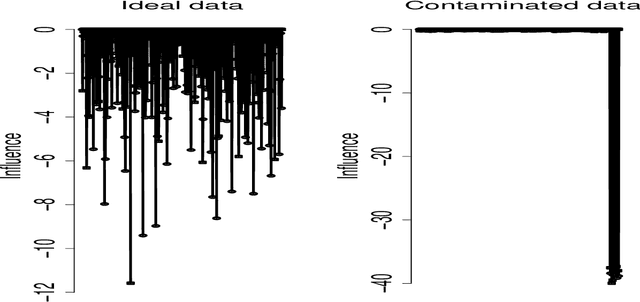


Imaging genetic research has essentially focused on discovering unique and co-association effects, but typically ignoring to identify outliers or atypical objects in genetic as well as non-genetics variables. Identifying significant outliers is an essential and challenging issue for imaging genetics and multiple sources data analysis. Therefore, we need to examine for transcription errors of identified outliers. First, we address the influence function (IF) of kernel mean element, kernel covariance operator, kernel cross-covariance operator, kernel canonical correlation analysis (kernel CCA) and multiple kernel CCA. Second, we propose an IF of multiple kernel CCA, which can be applied for more than two datasets. Third, we propose a visualization method to detect influential observations of multiple sources of data based on the IF of kernel CCA and multiple kernel CCA. Finally, the proposed methods are capable of analyzing outliers of subjects usually found in biomedical applications, in which the number of dimension is large. To examine the outliers, we use the stem-and-leaf display. Experiments on both synthesized and imaging genetics data (e.g., SNP, fMRI, and DNA methylation) demonstrate that the proposed visualization can be applied effectively.