 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCausal Queries from Observational Data in Biological Systems via Bayesian Networks: An Empirical Study in Small Networks
May 04, 2018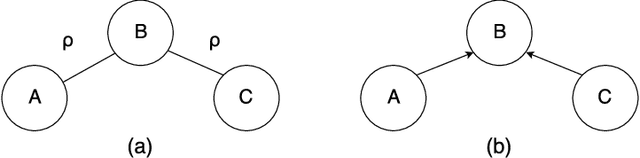
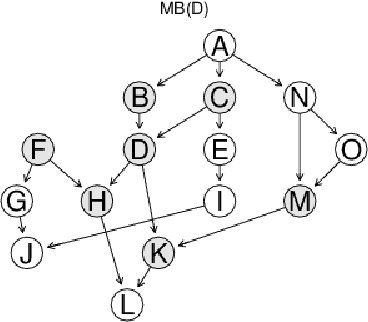
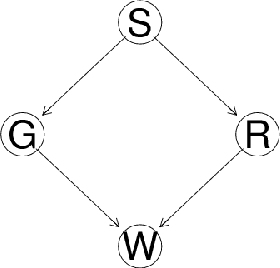
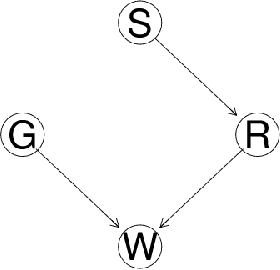
Biological networks are a very convenient modelling and visualisation tool to discover knowledge from modern high-throughput genomics and postgenomics data sets. Indeed, biological entities are not isolated, but are components of complex multi-level systems. We go one step further and advocate for the consideration of causal representations of the interactions in living systems.We present the causal formalism and bring it out in the context of biological networks, when the data is observational. We also discuss its ability to decipher the causal information flow as observed in gene expression. We also illustrate our exploration by experiments on small simulated networks as well as on a real biological data set.
Exact and approximate inference in graphical models: variable elimination and beyond
Mar 12, 2018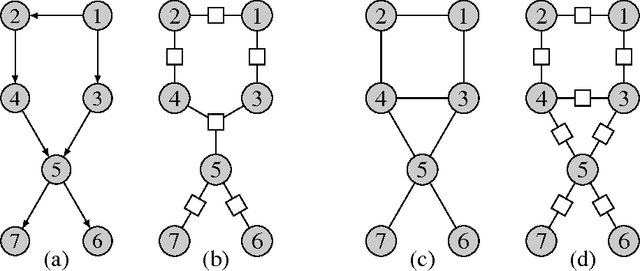

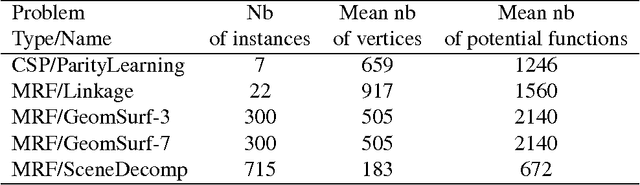
Probabilistic graphical models offer a powerful framework to account for the dependence structure between variables, which is represented as a graph. However, the dependence between variables may render inference tasks intractable. In this paper we review techniques exploiting the graph structure for exact inference, borrowed from optimisation and computer science. They are built on the principle of variable elimination whose complexity is dictated in an intricate way by the order in which variables are eliminated. The so-called treewidth of the graph characterises this algorithmic complexity: low-treewidth graphs can be processed efficiently. The first message that we illustrate is therefore the idea that for inference in graphical model, the number of variables is not the limiting factor, and it is worth checking for the treewidth before turning to approximate methods. We show how algorithms providing an upper bound of the treewidth can be exploited to derive a 'good' elimination order enabling to perform exact inference. The second message is that when the treewidth is too large, algorithms for approximate inference linked to the principle of variable elimination, such as loopy belief propagation and variational approaches, can lead to accurate results while being much less time consuming than Monte-Carlo approaches. We illustrate the techniques reviewed in this article on benchmarks of inference problems in genetic linkage analysis and computer vision, as well as on hidden variables restoration in coupled Hidden Markov Models.