 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGLUE: Generative Latent Unification of Expertise-Informed Engineering Models
Dec 22, 2025Engineering complex systems (aircraft, buildings, vehicles) requires accounting for geometric and performance couplings across subsystems. As generative models proliferate for specialized domains (wings, structures, engines), a key research gap is how to coordinate frozen, pre-trained submodels to generate full-system designs that are feasible, diverse, and high-performing. We introduce Generative Latent Unification of Expertise-Informed Engineering Models (GLUE), which orchestrates pre-trained, frozen subsystem generators while enforcing system-level feasibility, optimality, and diversity. We propose and benchmark (i) data-driven GLUE models trained on pre-generated system-level designs and (ii) a data-free GLUE model trained online on a differentiable geometry layer. On a UAV design problem with five coupling constraints, we find that data-driven approaches yield diverse, high-performing designs but require large datasets to satisfy constraints reliably. The data-free approach is competitive with Bayesian optimization and gradient-based optimization in performance and feasibility while training a full generative model in only 10 min on a RTX 4090 GPU, requiring more than two orders of magnitude fewer geometry evaluations and FLOPs than the data-driven method. Ablations focused on data-free training show that subsystem output continuity affects coordination, and equality constraints can trigger mode collapse unless mitigated. By integrating unmodified, domain-informed submodels into a modular generative workflow, this work provides a viable path for scaling generative design to complex, real-world engineering systems.
Assessing the trade-off between prediction accuracy and interpretability for topic modeling on energetic materials corpora
Jun 01, 2022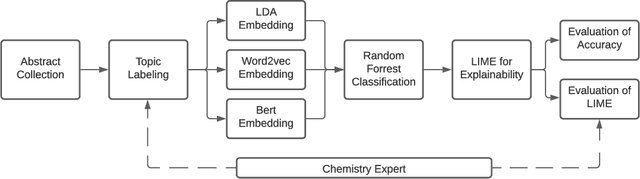
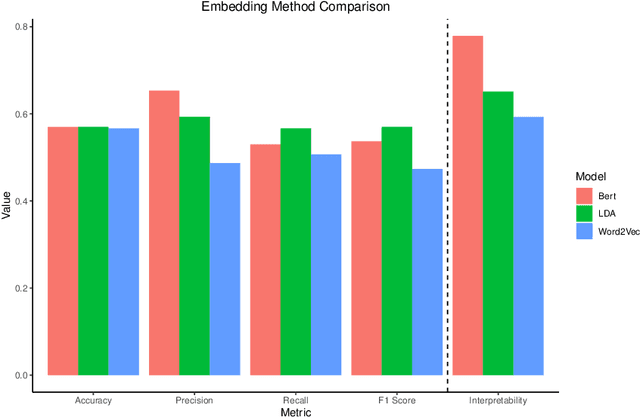

As the amount and variety of energetics research increases, machine aware topic identification is necessary to streamline future research pipelines. The makeup of an automatic topic identification process consists of creating document representations and performing classification. However, the implementation of these processes on energetics research imposes new challenges. Energetics datasets contain many scientific terms that are necessary to understand the context of a document but may require more complex document representations. Secondly, the predictions from classification must be understandable and trusted by the chemists within the pipeline. In this work, we study the trade-off between prediction accuracy and interpretability by implementing three document embedding methods that vary in computational complexity. With our accuracy results, we also introduce local interpretability model-agnostic explanations (LIME) of each prediction to provide a localized understanding of each prediction and to validate classifier decisions with our team of energetics experts. This study was carried out on a novel labeled energetics dataset created and validated by our team of energetics experts.
Deep learning for molecular generation and optimization - a review of the state of the art
Mar 11, 2019
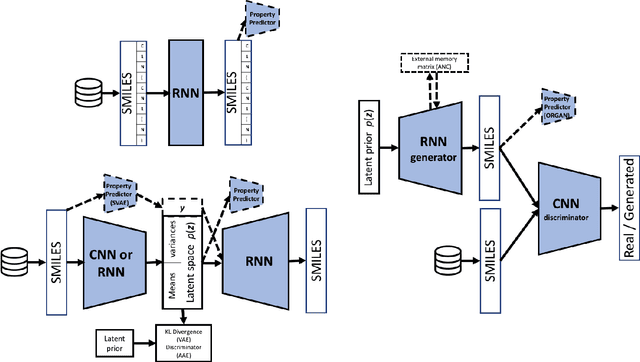
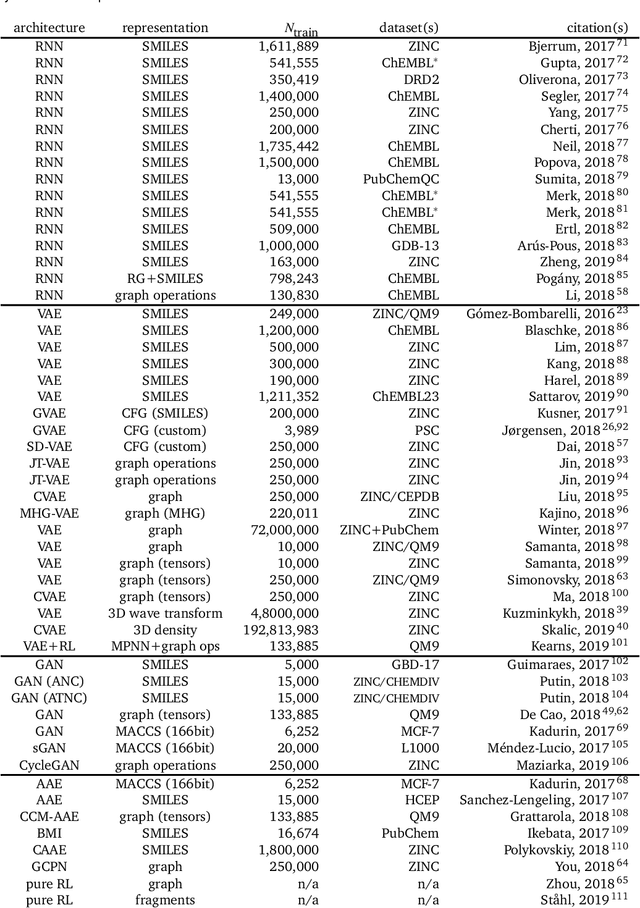
In the space of only a few years, deep generative modeling has revolutionized how we think of artificial creativity, yielding autonomous systems which produce original images, music, and text. Inspired by these successes, researchers are now applying deep generative modeling techniques to the generation and optimization of molecules - in our review we found 45 papers on the subject published in the past two years. These works point to a future where such systems will be used to generate lead molecules, greatly reducing resources spent downstream synthesizing and characterizing bad leads in the lab. In this review we survey the increasingly complex landscape of models and representation schemes that have been proposed. The four classes of techniques we describe are recursive neural networks, autoencoders, generative adversarial networks, and reinforcement learning. After first discussing some of the mathematical fundamentals of each technique, we draw high level connections and comparisons with other techniques and expose the pros and cons of each. Several important high level themes emerge as a result of this work, including the shift away from the SMILES string representation of molecules towards more sophisticated representations such as graph grammars and 3D representations, the importance of reward function design, the need for better standards for benchmarking and testing, and the benefits of adversarial training and reinforcement learning over maximum likelihood based training.
Using natural language processing techniques to extract information on the properties and functionalities of energetic materials from large text corpora
Mar 01, 2019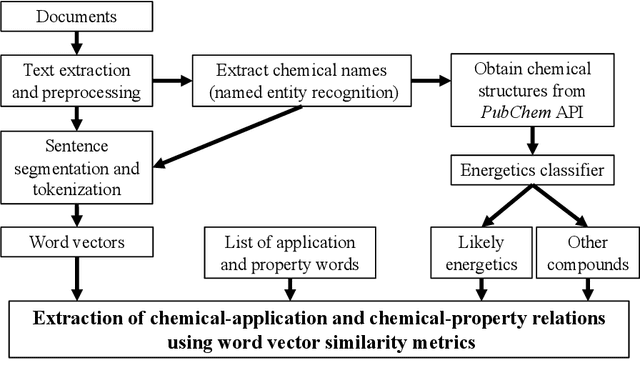
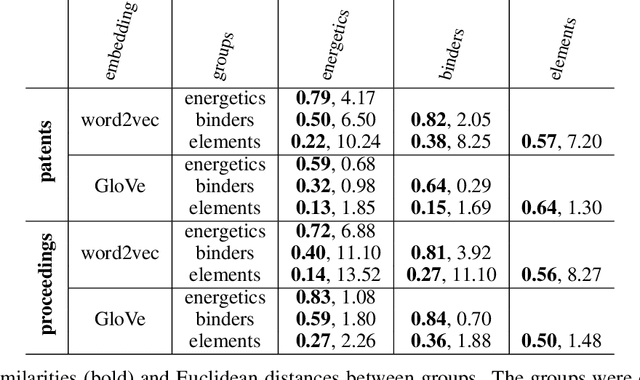
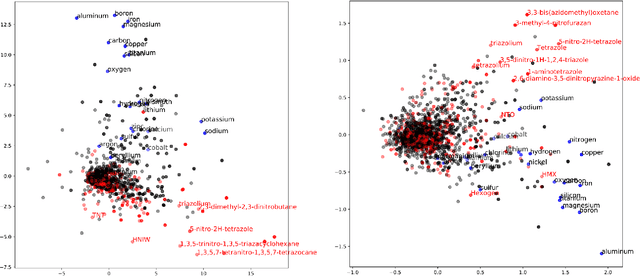

The number of scientific journal articles and reports being published about energetic materials every year is growing exponentially, and therefore extracting relevant information and actionable insights from the latest research is becoming a considerable challenge. In this work we explore how techniques from natural language processing and machine learning can be used to automatically extract chemical insights from large collections of documents. We first describe how to download and process documents from a variety of sources - journal articles, conference proceedings (including NTREM), the US Patent & Trademark Office, and the Defense Technical Information Center archive on archive.org. We present a custom NLP pipeline which uses open source NLP tools to identify the names of chemical compounds and relates them to function words ("underwater", "rocket", "pyrotechnic") and property words ("elastomer", "non-toxic"). After explaining how word embeddings work we compare the utility of two popular word embeddings - word2vec and GloVe. Chemical-chemical and chemical-application relationships are obtained by doing computations with word vectors. We show that word embeddings capture latent information about energetic materials, so that related materials appear close together in the word embedding space.
Independent Vector Analysis for Data Fusion Prior to Molecular Property Prediction with Machine Learning
Nov 01, 2018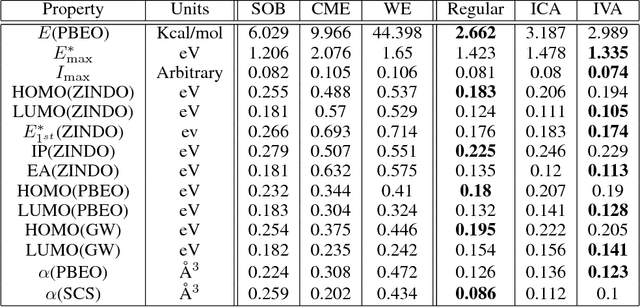
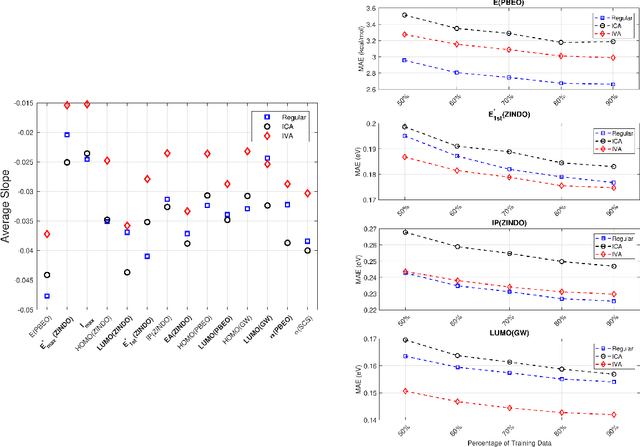
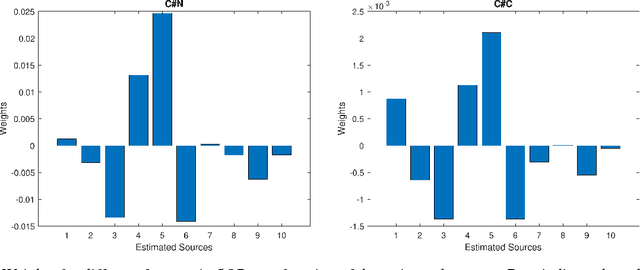
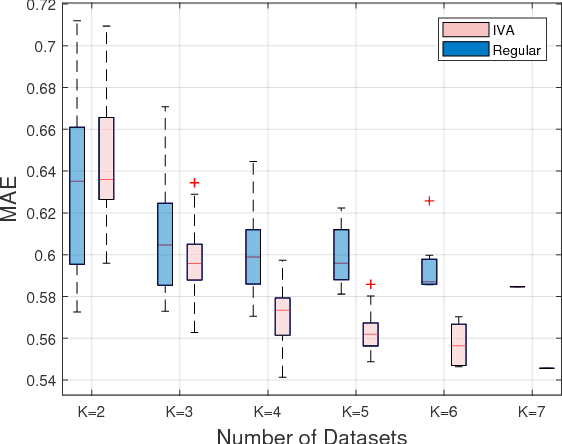
Due to its high computational speed and accuracy compared to ab-initio quantum chemistry and forcefield modeling, the prediction of molecular properties using machine learning has received great attention in the fields of materials design and drug discovery. A main ingredient required for machine learning is a training dataset consisting of molecular features\textemdash for example fingerprint bits, chemical descriptors, etc. that adequately characterize the corresponding molecules. However, choosing features for any application is highly non-trivial. No "universal" method for feature selection exists. In this work, we propose a data fusion framework that uses Independent Vector Analysis to exploit underlying complementary information contained in different molecular featurization methods, bringing us a step closer to automated feature generation. Our approach takes an arbitrary number of individual feature vectors and automatically generates a single, compact (low dimensional) set of molecular features that can be used to enhance the prediction performance of regression models. At the same time our methodology retains the possibility of interpreting the generated features to discover relationships between molecular structures and properties. We demonstrate this on the QM7b dataset for the prediction of several properties such as atomization energy, polarizability, frontier orbital eigenvalues, ionization potential, electron affinity, and excitation energies. In addition, we show how our method helps improve the prediction of experimental binding affinities for a set of human BACE-1 inhibitors. To encourage more widespread use of IVA we have developed the PyIVA Python package, an open source code which is available for download on Github.