 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge${\it Asparagus}$: A Toolkit for Autonomous, User-Guided Construction of Machine-Learned Potential Energy Surfaces
Jul 21, 2024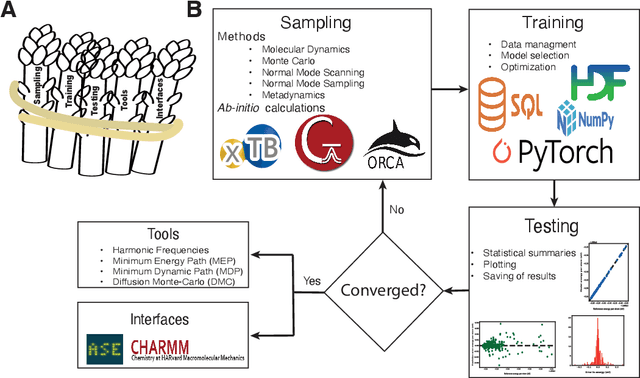



With the establishment of machine learning (ML) techniques in the scientific community, the construction of ML potential energy surfaces (ML-PES) has become a standard process in physics and chemistry. So far, improvements in the construction of ML-PES models have been conducted independently, creating an initial hurdle for new users to overcome and complicating the reproducibility of results. Aiming to reduce the bar for the extensive use of ML-PES, we introduce ${\it Asparagus}$, a software package encompassing the different parts into one coherent implementation that allows an autonomous, user-guided construction of ML-PES models. ${\it Asparagus}$ combines capabilities of initial data sampling with interfaces to ${\it ab initio}$ calculation programs, ML model training, as well as model evaluation and its application within other codes such as ASE or CHARMM. The functionalities of the code are illustrated in different examples, including the dynamics of small molecules, the representation of reactive potentials in organometallic compounds, and atom diffusion on periodic surface structures. The modular framework of ${\it Asparagus}$ is designed to allow simple implementations of further ML-related methods and models to provide constant user-friendly access to state-of-the-art ML techniques.
Outlier-Detection for Reactive Machine Learned Potential Energy Surfaces
Feb 27, 2024Uncertainty quantification (UQ) to detect samples with large expected errors (outliers) is applied to reactive molecular potential energy surfaces (PESs). Three methods - Ensembles, Deep Evidential Regression (DER), and Gaussian Mixture Models (GMM) - were applied to the H-transfer reaction between ${\it syn-}$Criegee and vinyl hydroxyperoxide. The results indicate that ensemble models provide the best results for detecting outliers, followed by GMM. For example, from a pool of 1000 structures with the largest uncertainty, the detection quality for outliers is $\sim 90$ \% and $\sim 50$ \%, respectively, if 25 or 1000 structures with large errors are sought. On the contrary, the limitations of the statistical assumptions of DER greatly impacted its prediction capabilities. Finally, a structure-based indicator was found to be correlated with large average error, which may help to rapidly classify new structures into those that provide an advantage for refining the neural network.
Uncertainty quantification for predictions of atomistic neural networks
Jul 21, 2022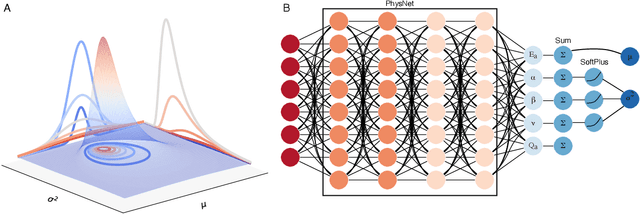
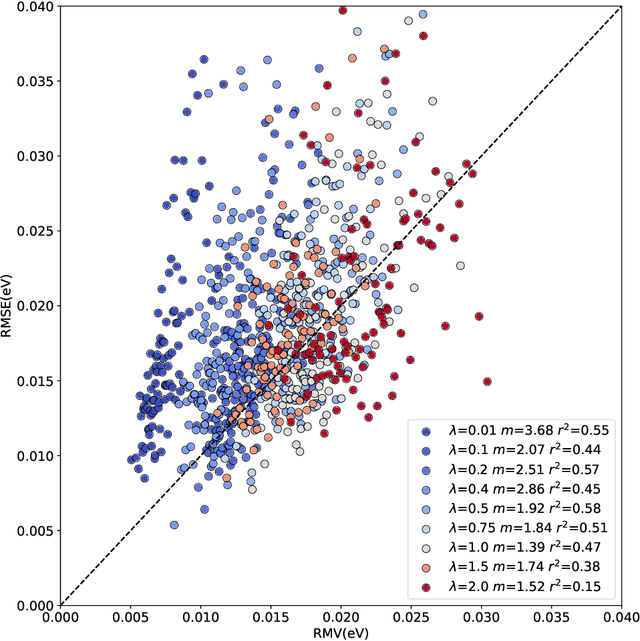
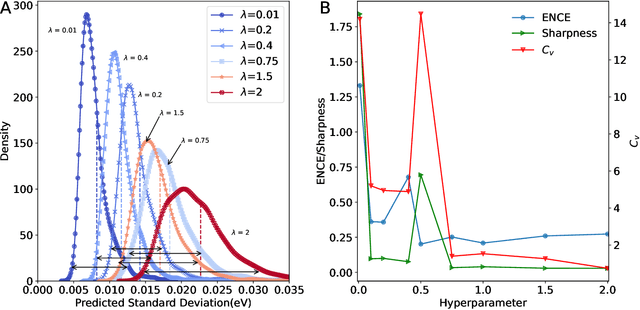
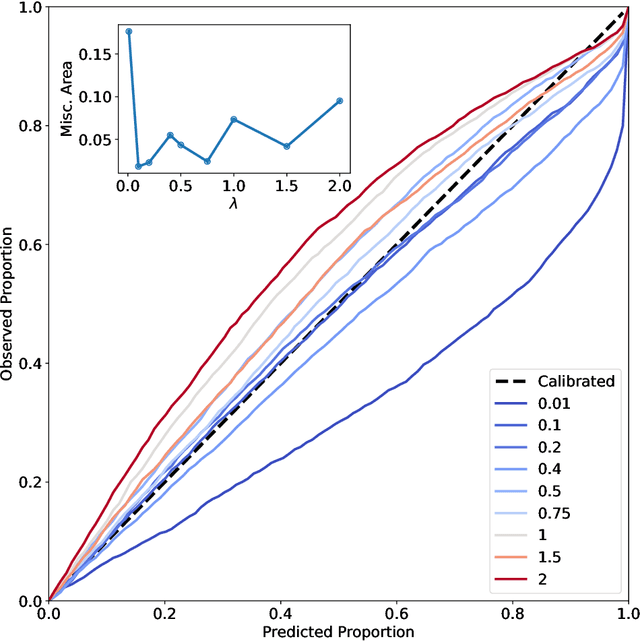
The value of uncertainty quantification on predictions for trained neural networks (NNs) on quantum chemical reference data is quantitatively explored. For this, the architecture of the PhysNet NN was suitably modified and the resulting model was evaluated with different metrics to quantify calibration, quality of predictions, and whether prediction error and the predicted uncertainty can be correlated. The results from training on the QM9 database and evaluating data from the test set within and outside the distribution indicate that error and uncertainty are not linearly related. The results clarify that noise and redundancy complicate property prediction for molecules even in cases for which changes - e.g. double bond migration in two otherwise identical molecules - are small. The model was then applied to a real database of tautomerization reactions. Analysis of the distance between members in feature space combined with other parameters shows that redundant information in the training dataset can lead to large variances and small errors whereas the presence of similar but unspecific information returns large errors but small variances. This was, e.g., observed for nitro-containing aliphatic chains for which predictions were difficult although the training set contained several examples for nitro groups bound to aromatic molecules. This underlines the importance of the composition of the training data and provides chemical insight into how this affects the prediction capabilities of a ML model. Finally, the approach put forward can be used for information-based improvement of chemical databases for target applications through active learning optimization.