 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSynCraft: Guiding Large Language Models to Predict Edit Sequences for Molecular Synthesizability Optimization
Dec 23, 2025Generative artificial intelligence has revolutionized the exploration of chemical space, yet a critical bottleneck remains that a substantial fraction of generated molecules is synthetically inaccessible. Current solutions, such as post-hoc filtering or projection-based methods, often compromise structural novelty or disrupt key pharmacophores by forcing molecules into pre-defined synthetic templates. Herein, we introduce SynCraft, a reasoning-based framework that reframes synthesizability optimization not as a sequence translation task, but as a precise structural editing problem. Leveraging the emergent reasoning capabilities of Large Language Models, SynCraft navigates the "synthesis cliff" where minimal structural modifications yield significant gains in synthetic feasibility. By predicting executable sequences of atom-level edits rather than generating SMILES strings directly, SynCraft circumvents the syntactic fragility of LLMs while harnessing their chemical intuition. Extensive benchmarks demonstrate that SynCraft outperforms state-of-the-art baselines in generating synthesizable analogs with high structural fidelity. Furthermore, through interaction-aware prompting, SynCraft successfully replicates expert medicinal chemistry intuition in editing PLK1 inhibitors and rescuing high-scoring but previously discarded RIPK1 candidates in previous molecular generation literatures.
MolMiner: You only look once for chemical structure recognition
May 23, 2022
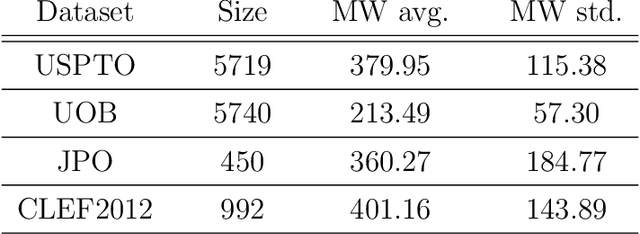
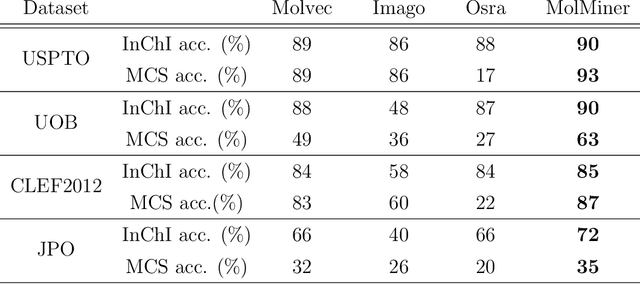
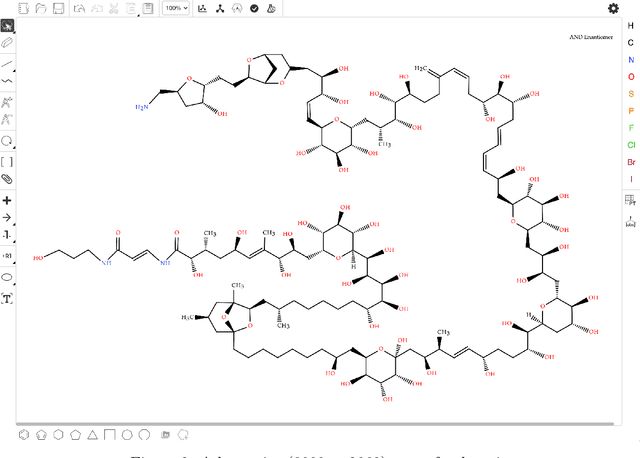
Molecular structures are always depicted as 2D printed form in scientific documents like journal papers and patents. However, these 2D depictions are not machine-readable. Due to a backlog of decades and an increasing amount of these printed literature, there is a high demand for the translation of printed depictions into machine-readable formats, which is known as Optical Chemical Structure Recognition (OCSR). Most OCSR systems developed over the last three decades follow a rule-based approach where the key step of vectorization of the depiction is based on the interpretation of vectors and nodes as bonds and atoms. Here, we present a practical software MolMiner, which is primarily built up using deep neural networks originally developed for semantic segmentation and object detection to recognize atom and bond elements from documents. These recognized elements can be easily connected as a molecular graph with distance-based construction algorithm. We carefully evaluate our software on four benchmark datasets with the state-of-the-art performance. Various real application scenarios are also tested, yielding satisfactory outcomes. The free download links of Mac and Windows versions are available: Mac: https://molminer-cdn.iipharma.cn/pharma-mind/artifact/latest/mac/PharmaMind-mac-latest-setup.dmg and Windows: https://molminer-cdn.iipharma.cn/pharma-mind/artifact/latest/win/PharmaMind-win-latest-setup.exe
Learning to design drug-like molecules in three-dimensional space using deep generative models
Apr 17, 2021
Recently, deep generative models for molecular graphs are gaining more and more attention in the field of de novo drug design. A variety of models have been developed to generate topological structures of drug-like molecules, but explorations in generating three-dimensional structures are still limited. Existing methods have either focused on low molecular weight compounds without considering drug-likeness or generate 3D structures indirectly using atom density maps. In this work, we introduce Ligand Neural Network (L-Net), a novel graph generative model for designing drug-like molecules with high-quality 3D structures. L-Net directly outputs the topological and 3D structure of molecules (including hydrogen atoms), without the need for additional atom placement or bond order inference algorithm. The architecture of L-Net is specifically optimized for drug-like molecules, and a set of metrics is assembled to comprehensively evaluate its performance. The results show that L-Net is capable of generating chemically correct, conformationally valid, and highly druglike molecules. Finally, to demonstrate its potential in structure-based molecular design, we combine L-Net with MCTS and test its ability to generate potential inhibitors targeting ABL1 kinase.