 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning to design drug-like molecules in three-dimensional space using deep generative models
Paper and Code
Apr 17, 2021
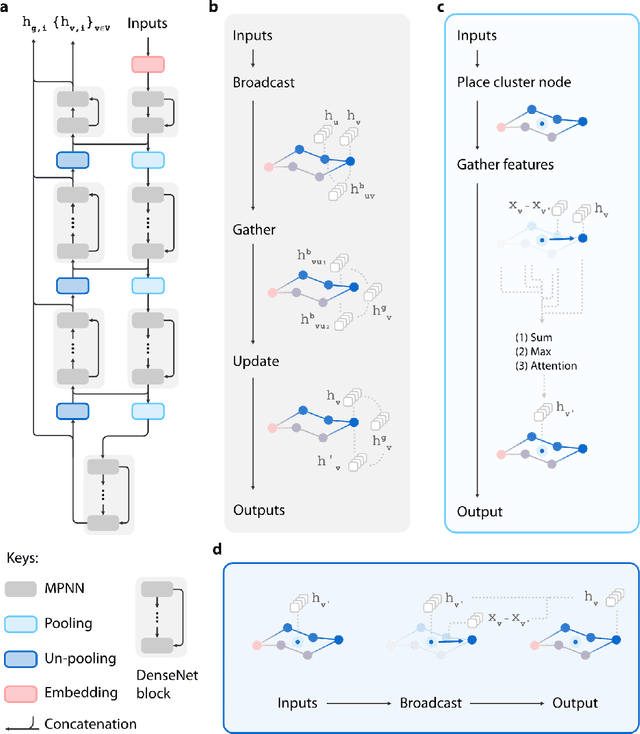
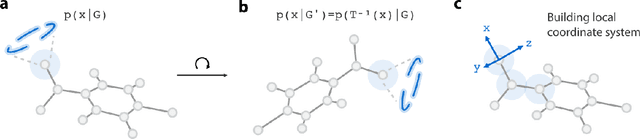
Recently, deep generative models for molecular graphs are gaining more and more attention in the field of de novo drug design. A variety of models have been developed to generate topological structures of drug-like molecules, but explorations in generating three-dimensional structures are still limited. Existing methods have either focused on low molecular weight compounds without considering drug-likeness or generate 3D structures indirectly using atom density maps. In this work, we introduce Ligand Neural Network (L-Net), a novel graph generative model for designing drug-like molecules with high-quality 3D structures. L-Net directly outputs the topological and 3D structure of molecules (including hydrogen atoms), without the need for additional atom placement or bond order inference algorithm. The architecture of L-Net is specifically optimized for drug-like molecules, and a set of metrics is assembled to comprehensively evaluate its performance. The results show that L-Net is capable of generating chemically correct, conformationally valid, and highly druglike molecules. Finally, to demonstrate its potential in structure-based molecular design, we combine L-Net with MCTS and test its ability to generate potential inhibitors targeting ABL1 kinase.