 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTargeting the partition function of chemically disordered materials with a generative approach based on inverse variational autoencoders
Sep 10, 2024Computing atomic-scale properties of chemically disordered materials requires an efficient exploration of their vast configuration space. Traditional approaches such as Monte Carlo or Special Quasirandom Structures either entail sampling an excessive amount of configurations or do not ensure that the configuration space has been properly covered. In this work, we propose a novel approach where generative machine learning is used to yield a representative set of configurations for accurate property evaluation and provide accurate estimations of atomic-scale properties with minimal computational cost. Our method employs a specific type of variational autoencoder with inverse roles for the encoder and decoder, enabling the application of an unsupervised active learning scheme that does not require any initial training database. The model iteratively generates configuration batches, whose properties are computed with conventional atomic-scale methods. These results are then fed back into the model to estimate the partition function, repeating the process until convergence. We illustrate our approach by computing point-defect formation energies and concentrations in (U, Pu)O2 mixed-oxide fuels. In addition, the ML model provides valuable insights into the physical factors influencing the target property. Our method is generally applicable to explore other properties, such as atomic-scale diffusion coefficients, in ideally or non-ideally disordered materials like high-entropy alloys.
Targetin the partition function of chemically disordered materials with a generative approach based on inverse variational autoencoders
Aug 27, 2024Computing atomic-scale properties of chemically disordered materials requires an efficient exploration of their vast configuration space. Traditional approaches such as Monte Carlo or Special Quasirandom Structures either entail sampling an excessive amount of configurations or do not ensure that the configuration space has been properly covered. In this work, we propose a novel approach where generative machine learning is used to yield a representative set of configurations for accurate property evaluation and provide accurate estimations of atomic-scale properties with minimal computational cost. Our method employs a specific type of variational autoencoder with inverse roles for the encoder and decoder, enabling the application of an unsupervised active learning scheme that does not require any initial training database. The model iteratively generates configuration batches, whose properties are computed with conventional atomic-scale methods. These results are then fed back into the model to estimate the partition function, repeating the process until convergence. We illustrate our approach by computing point-defect formation energies and concentrations in (U, Pu)O2 mixed-oxide fuels. In addition, the ML model provides valuable insights into the physical factors influencing the target property. Our method is generally applicable to explore other properties, such as atomic-scale diffusion coefficients, in ideally or non-ideally disordered materials like high-entropy alloys.
Smart energy models for atomistic simulations using a DFT-driven multifidelity approach
Aug 28, 2018
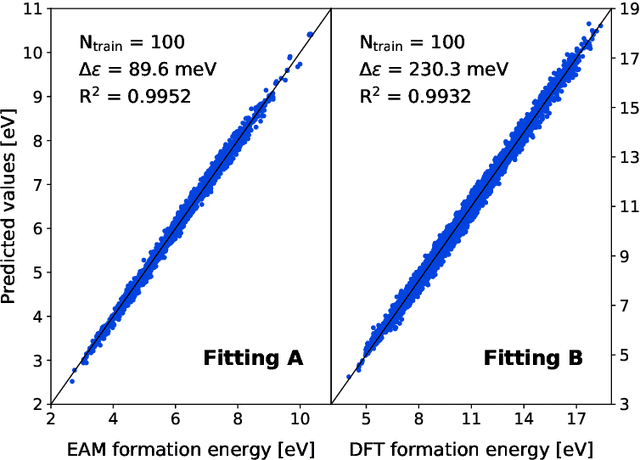

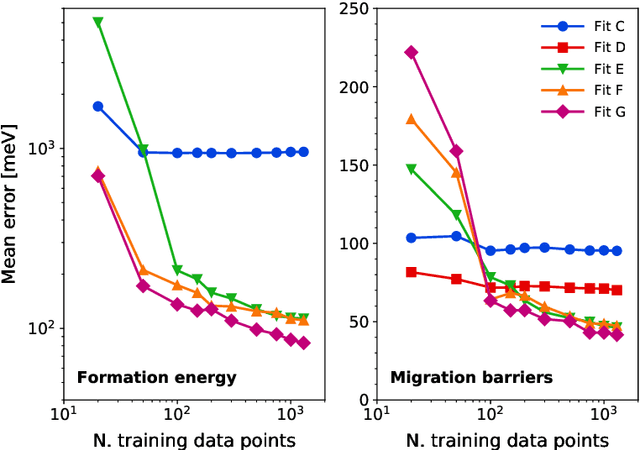
The reliability of atomistic simulations depends on the quality of the underlying energy models providing the source of physical information, for instance for the calculation of migration barriers in atomistic Kinetic Monte Carlo simulations. Accurate (high-fidelity) methods are often available, but since they are usually computationally expensive, they must be replaced by less accurate (low-fidelity) models that introduce some degrees of approximation. Machine-learning techniques such as artificial neural networks are usually employed to work around this limitation and extract the needed parameters from large databases of high-fidelity data, but the latter are often computationally expensive to produce. This work introduces an alternative method based on the multifidelity approach, where correlations between high-fidelity and low-fidelity outputs are exploited to make an educated guess of the high-fidelity outcome based only on quick low-fidelity estimations, hence without the need of running full expensive high-fidelity calculations. With respect to neural networks, this approach is expected to require less training data because of the lower amount of fitting parameters involved. The method is tested on the prediction of ab initio formation and migration energies of vacancy diffusion in iron-copper alloys, and compared with the neural networks trained on the same database.