 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMANAR: Memory-augmented Attention with Navigational Abstract Conceptual Representation
Mar 19, 2026MANAR (Memory-augmented Attention with Navigational Abstract Conceptual Representation), contextualization layer generalizes standard multi-head attention (MHA) by instantiating the principles of Global Workspace Theory (GWT). While MHA enables unconstrained all-to-all communication, it lacks the functional bottleneck and global integration mechanisms hypothesized in cognitive models of consciousness. MANAR addresses this by implementing a central workspace through a trainable memory of abstract concepts and an Abstract Conceptual Representation (ACR). The architecture follows a two-stage logic that maps directly to GWT mechanics: (i) an integration phase, where retrieved memory concepts converge to form a collective "mental image" (the ACR) based on input stimuli; and (ii) a broadcasting phase, where this global state navigates and informs the contextualization of individual local tokens. We demonstrate that efficient linear-time scaling is a fundamental architectural byproduct of instantiating GWT functional bottleneck, as routing global information through a constant-sized ACR resolves the quadratic complexity inherent in standard attention. MANAR is a compatible re-parameterization of MHA with identical semantic roles for its projections, enabling knowledge transfer from pretrained transformers via weight-copy and thus overcoming the adoption barriers of structurally incompatible linear-time alternatives. MANAR enables non-convex contextualization, synthesizing representations that provably lie outside the convex hull of input tokens - a mathematical reflection of the creative synthesis described in GWT. Empirical evaluations confirm that MANAR matches or exceeds strong baselines across language (GLUE score of 85.1), vision (83.9% ImageNet-1K), and speech (2.7% WER on LibriSpeech), positioning it as an efficient and expressive alternative to quadratic attention.
ClaPIM: Scalable Sequence CLAssification using Processing-In-Memory
Feb 16, 2023DNA sequence classification is a fundamental task in computational biology with vast implications for applications such as disease prevention and drug design. Therefore, fast high-quality sequence classifiers are significantly important. This paper introduces ClaPIM, a scalable DNA sequence classification architecture based on the emerging concept of hybrid in-crossbar and near-crossbar memristive processing-in-memory (PIM). We enable efficient and high-quality classification by uniting the filter and search stages within a single algorithm. Specifically, we propose a custom filtering technique that drastically narrows the search space and a search approach that facilitates approximate string matching through a distance function. ClaPIM is the first PIM architecture for scalable approximate string matching that benefits from the high density of memristive crossbar arrays and the massive computational parallelism of PIM. Compared with Kraken2, a state-of-the-art software classifier, ClaPIM provides significantly higher classification quality (up to 20x improvement in F1 score) and also demonstrates a 1.8x throughput improvement. Compared with EDAM, a recently-proposed SRAM-based accelerator that is restricted to small datasets, we observe both a 30.4x improvement in normalized throughput per area and a 7% increase in classification precision.
CoViT: Real-time phylogenetics for the SARS-CoV-2 pandemic using Vision Transformers
Aug 09, 2022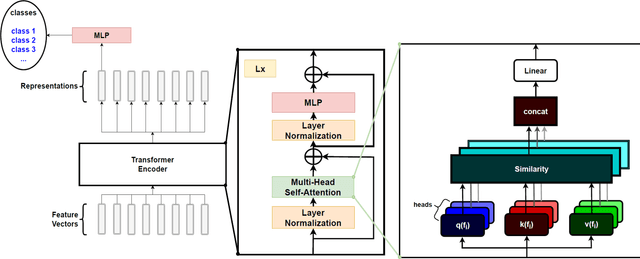
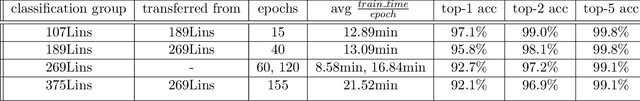
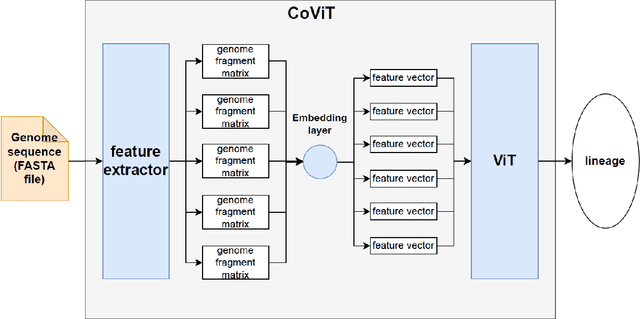

Real-time viral genome detection, taxonomic classification and phylogenetic analysis are critical for efficient tracking and control of viral pandemics such as Covid-19. However, the unprecedented and still growing amounts of viral genome data create a computational bottleneck, which effectively prevents the real-time pandemic tracking. We are attempting to alleviate this bottleneck by modifying and applying Vision Transformer, a recently developed neural network model for image recognition, to taxonomic classification and placement of viral genomes, such as SARS-CoV-2. Our solution, CoViT, places newly acquired samples onto the tree of SARS-CoV-2 lineages. One of the two potential placements returned by CoVit is the true one with the probability of 99.0%. The probability of the correct placement to be found among five potential placements generated by CoViT is 99.8%. The placement time is 1.45ms per individual genome running on NVIDIAs GeForce RTX 2080 Ti GPU. We make CoViT available to research community through GitHub: https://github.com/zuherJahshan/covit.