 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSpinMultiNet: Neural Network Potential Incorporating Spin Degrees of Freedom with Multi-Task Learning
Sep 05, 2024
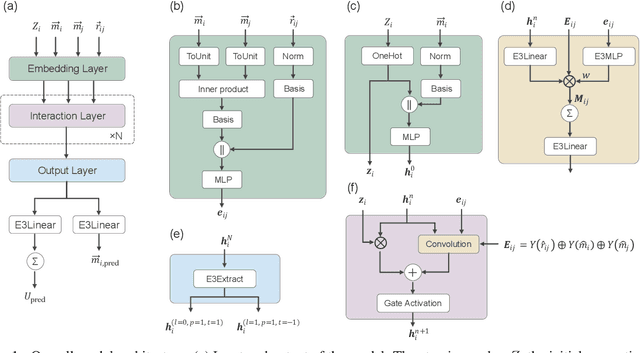

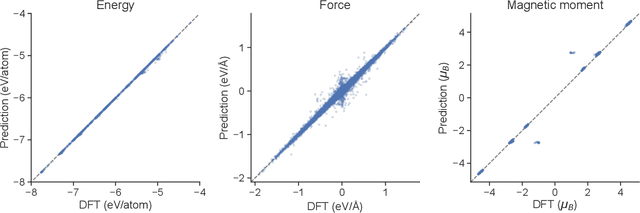
Neural Network Potentials (NNPs) have attracted significant attention as a method for accelerating density functional theory (DFT) calculations. However, conventional NNP models typically do not incorporate spin degrees of freedom, limiting their applicability to systems where spin states critically influence material properties, such as transition metal oxides. This study introduces SpinMultiNet, a novel NNP model that integrates spin degrees of freedom through multi-task learning. SpinMultiNet achieves accurate predictions without relying on correct spin values obtained from DFT calculations. Instead, it utilizes initial spin estimates as input and leverages multi-task learning to optimize the spin latent representation while maintaining both $E(3)$ and time-reversal equivariance. Validation on a dataset of transition metal oxides demonstrates the high predictive accuracy of SpinMultiNet. The model successfully reproduces the energy ordering of stable spin configurations originating from superexchange interactions and accurately captures the rhombohedral distortion of the rocksalt structure. These results pave the way for new possibilities in materials simulations that consider spin degrees of freedom, promising future applications in large-scale simulations of various material systems, including magnetic materials.
Shotgun crystal structure prediction using machine-learned formation energies
May 07, 2023Stable or metastable crystal structures of assembled atoms can be predicted by finding the global or local minima of the energy surface with respect to the atomic configurations. Generally, this requires repeated first-principles energy calculations that are impractical for large systems, such as those containing more than 30 atoms in the unit cell. Here, we have made significant progress in solving the crystal structure prediction problem with a simple but powerful machine-learning workflow; using a machine-learning surrogate for first-principles energy calculations, we performed non-iterative, single-shot screening using a large library of virtually created crystal structures. The present method relies on two key technical components: transfer learning, which enables a highly accurate energy prediction of pre-relaxed crystalline states given only a small set of training samples from first-principles calculations, and generative models to create promising and diverse crystal structures for screening. Here, first-principles calculations were performed only to generate the training samples, and for the optimization of a dozen or fewer finally narrowed-down crystal structures. Our shotgun method was more than 5--10 times less computationally demanding and achieved an outstanding prediction accuracy that was 2--6 times higher than that of the conventional methods that rely heavily on iterative first-principles calculations.