 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBidimensional linked matrix factorization for pan-omics pan-cancer analysis
Feb 07, 2020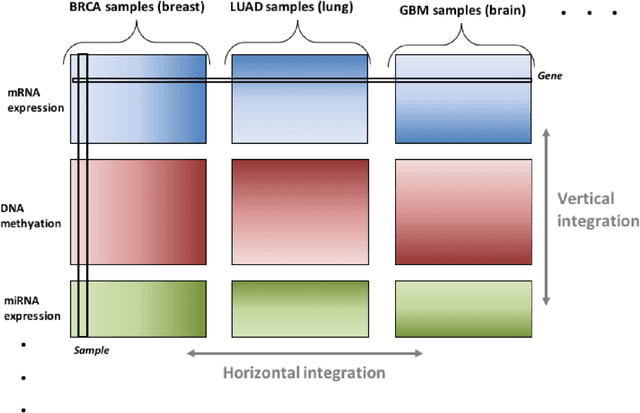
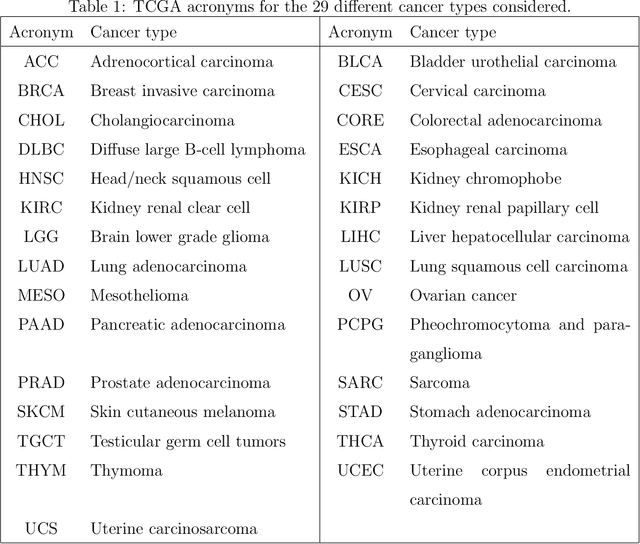
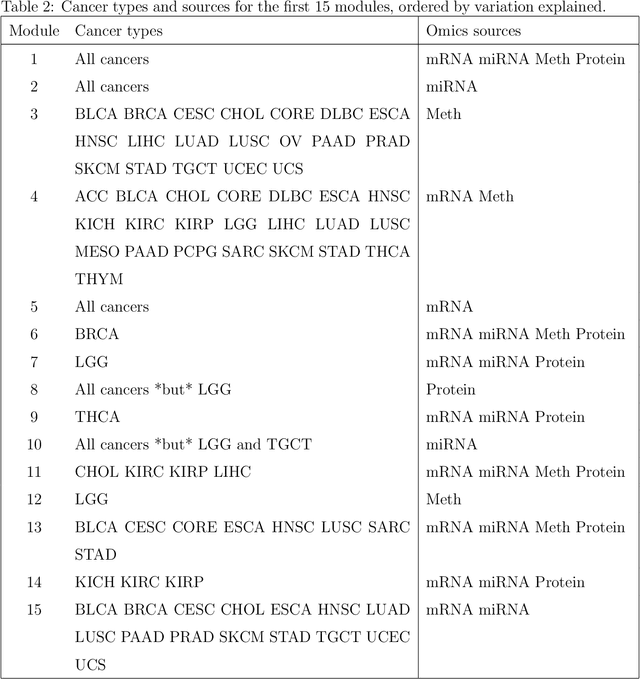
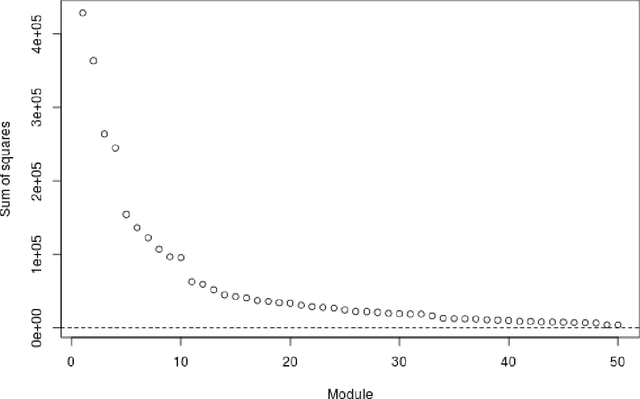
Several modern applications require the integration of multiple large data matrices that have shared rows and/or columns. For example, cancer studies that integrate multiple omics platforms across multiple types of cancer, pan-omics pan-cancer analysis, have extended our knowledge of molecular heterogenity beyond what was observed in single tumor and single platform studies. However, these studies have been limited by available statistical methodology. We propose a flexible approach to the simultaneous factorization and decomposition of variation across such bidimensionally linked matrices, BIDIFAC+. This decomposes variation into a series of low-rank components that may be shared across any number of row sets (e.g., omics platforms) or column sets (e.g., cancer types). This builds on a growing literature for the factorization and decomposition of linked matrices, which has primarily focused on multiple matrices that are linked in one dimension (rows or columns) only. Our objective function extends nuclear norm penalization, is motivated by random matrix theory, gives an identifiable decomposition under relatively mild conditions, and can be shown to give the mode of a Bayesian posterior distribution. We apply BIDIFAC+ to pan-omics pan-cancer data from TCGA, identifying shared and specific modes of variability across 4 different omics platforms and 29 different cancer types.
Joint and individual variation explained (JIVE) for integrated analysis of multiple data types
May 28, 2013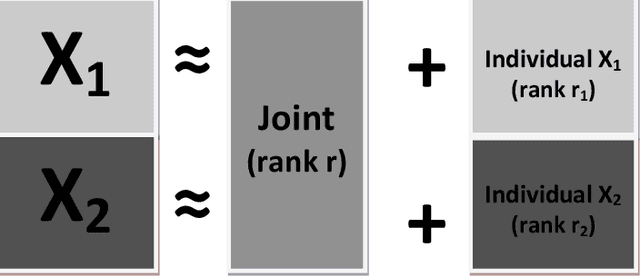
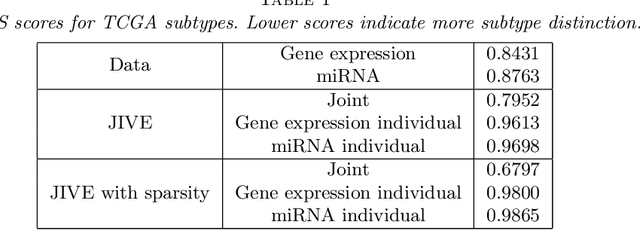
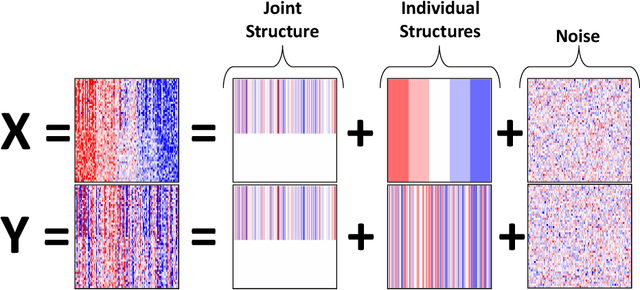
Research in several fields now requires the analysis of data sets in which multiple high-dimensional types of data are available for a common set of objects. In particular, The Cancer Genome Atlas (TCGA) includes data from several diverse genomic technologies on the same cancerous tumor samples. In this paper we introduce Joint and Individual Variation Explained (JIVE), a general decomposition of variation for the integrated analysis of such data sets. The decomposition consists of three terms: a low-rank approximation capturing joint variation across data types, low-rank approximations for structured variation individual to each data type, and residual noise. JIVE quantifies the amount of joint variation between data types, reduces the dimensionality of the data and provides new directions for the visual exploration of joint and individual structures. The proposed method represents an extension of Principal Component Analysis and has clear advantages over popular two-block methods such as Canonical Correlation Analysis and Partial Least Squares. A JIVE analysis of gene expression and miRNA data on Glioblastoma Multiforme tumor samples reveals gene-miRNA associations and provides better characterization of tumor types. Data and software are available at https://genome.unc.edu/jive/
* Published in at http://dx.doi.org/10.1214/12-AOAS597 the Annals of Applied Statistics (http://www.imstat.org/aoas/) by the Institute of Mathematical Statistics (http://www.imstat.org)